Metastasis is perhaps the most common reason for treatment failure in cancer patients, as well as the leading cause of cancer-related death. Mitogen-activated protein kinase (MAPK) are serine/threonine-protein kinases that can be activated by a variety of extracellular stimuli including growth factors, cytokines, insulin, environmental factors, and oxidative and genotoxic stress. It is becoming increasingly clear that MAPKs are involved in all the steps required for hyperproliferating cells to develop into metastatic tumors. However, we are currently lacking in vivo data to fully understand how MAPK signaling pathways can affect the progression of metastatic disease.
- cancer
- c-JUN N-terminal kinase (JNK)
- extracellular signal-regulated kinase (ERK)
- metastasis
- mitogen-activated kinases (MAPKs)
1. Molecular Basis of Metastasis
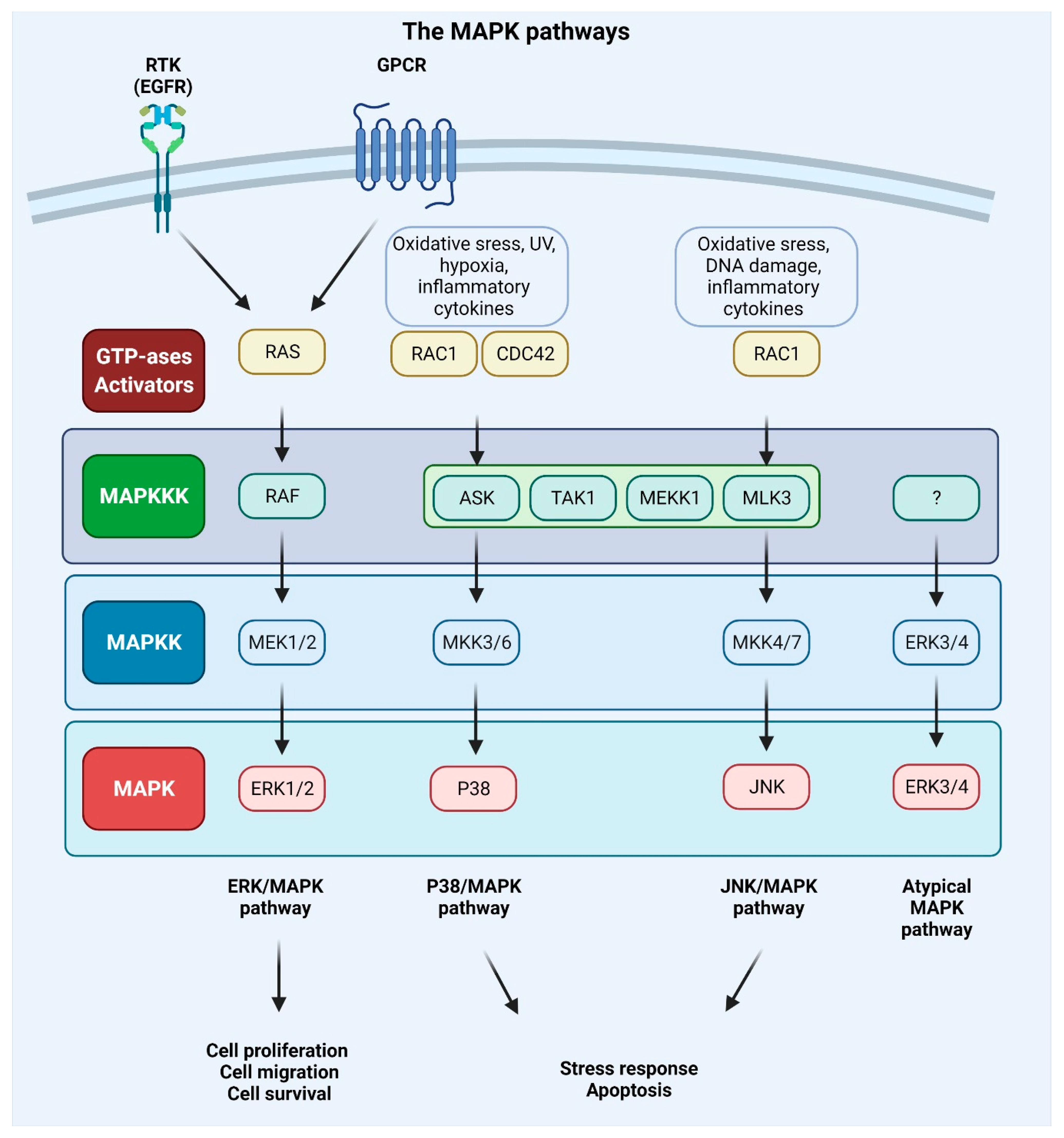
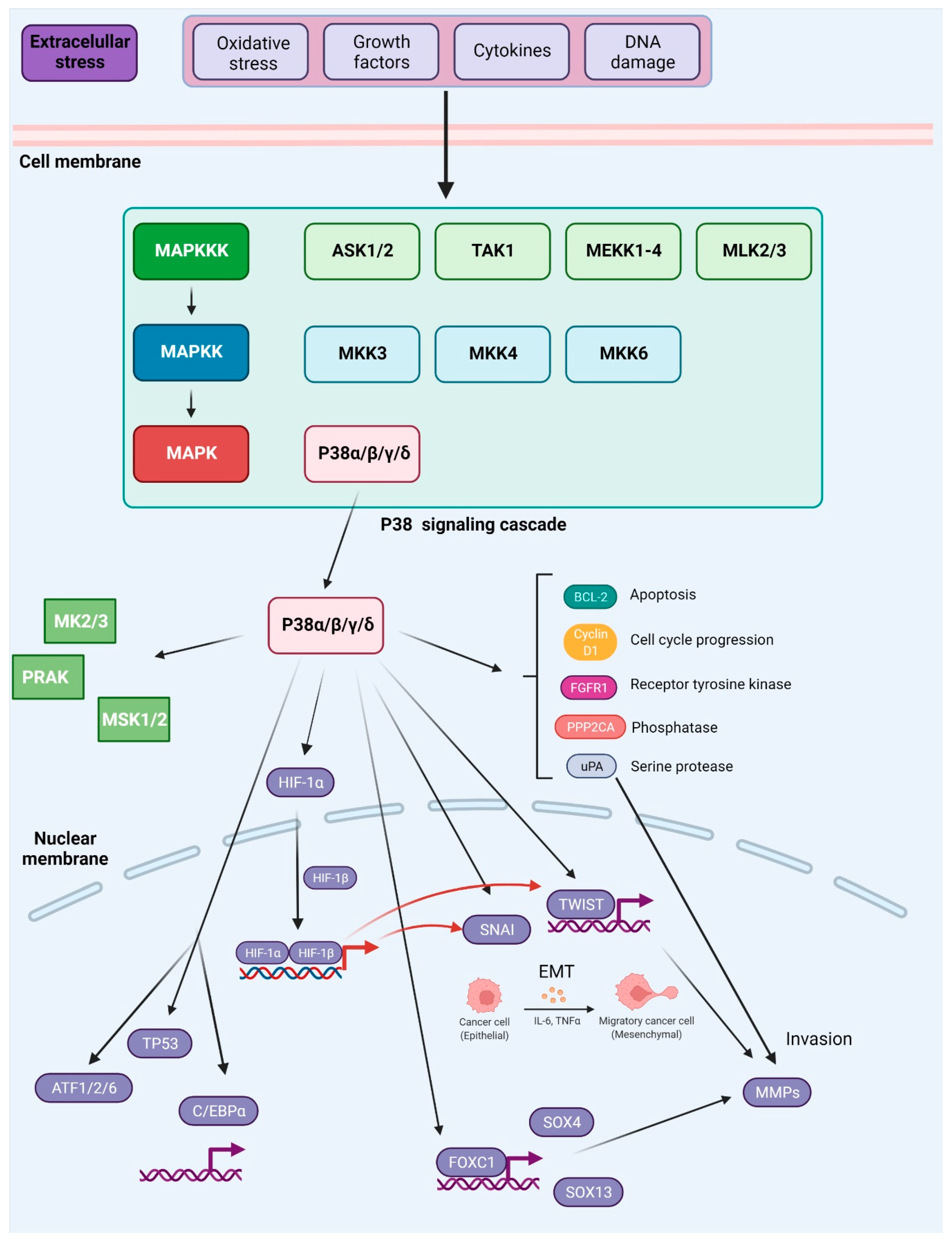
2. The MAPK Pathway in Metastasis
This entry is adapted from the peer-reviewed paper 10.3390/ijms23073847
References
- Nguyen, D.X.; Massagué, J. Genetic determinants of cancer metastasis. Nat. Rev. Genet. 2007, 8, 341–352.
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28.
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019, 21, 102–112.
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890.
- Zhang, J.; Ma, L. MicroRNA control of epithelial-mesenchymal transition and metastasis. Cancer Metastasis Rev. 2012, 31, 653–662.
- Xu, Q.; Deng, F.; Qin, Y.; Zhao, Z.; Wu, Z.; Xing, Z.; Ji, A.; Wang, Q.J. Long non-coding RNA regulation of epithelial-mesenchymal transition in cancer metastasis. Cell Death Dis. 2016, 7, e2254.
- Asl, E.R.; Amini, M.; Najafi, S.; Mansoori, B.; Mokhtarzadeh, A.; Mohammadi, A.; Lotfinejad, P.; Bagheri, M.; Shirjang, S.; Lotfi, Z.; et al. Interplay between MAPK/ERK signaling pathway and MicroRNAs: A crucial mechanism regulating cancer cell metabolism and tumor progression. Life Sci. 2021, 278, 119499.
- Magnelli, L.; Schiavone, N.; Staderini, F.; Biagioni, A.; Papucci, L. MAP Kinases Pathways in Gastric Cancer. Int. J. Mol. Sci. 2020, 21, 2893.
- Soleimani, A.; Rahmani, F.; Saeedi, N.; Ghaffarian, R.; Khazaei, M.; Ferns, G.A.; Avan, A.; Hassanian, S.M. The potential role of regulatory microRNAs of RAS/MAPK signaling pathway in the pathogenesis of colorectal cancer. J. Cell. Biochem. 2019, 120, 19245–19253.
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18.
- Paňková, K.; Rösel, D.; Novotný, M.; Brábek, J. The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells. Cell. Mol. Life Sci. 2010, 67, 63–71.
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196.
- Guan, X. Cancer metastases: Challenges and opportunities. Acta Pharm. Sin. B 2015, 5, 402–418.
- Winer, A.; Adams, S.; Mignatti, P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures into Future Successes. Mol. Cancer Ther. 2018, 17, 1147–1155.
- Deryugina, E.I.; Quigley, J.P. Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biol. 2015, 44, 94–112.
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzyme Inhib. Med. Chem. 2016, 31, 177–183.
- Amano, S.; Akutsu, N.; Matsunaga, Y.; Nishiyama, T.; Champliaud, M.F.; Burgeson, R.E.; Adachi, E. Importance of balance between extracellular matrix synthesis and degradation in basement membrane formation. Exp. Cell Res. 2001, 271, 249–262.
- Rundhaug, J.E. Matrix metalloproteinases and angiogenesis. J. Cell. Mol. Med. 2005, 9, 267–285.
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370.
- McQuibban, G.A.; Gong, J.H.; Tam, E.M.; McCulloch, C.A.; Clark-Lewis, I.; Overall, C.M. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science 2000, 289, 1202–1206.
- Godefroy, E.; Manches, O.; Dréno, B.; Hochman, T.; Rolnitzky, L.; Labarrière, N.; Guilloux, Y.; Goldberg, J.; Jotereau, F.; Bhardwaj, N. Matrix metalloproteinase-2 conditions human dendritic cells to prime inflammatory T(H)2 cells via an IL-12- and OX40L-dependent pathway. Cancer Cell 2011, 19, 333–346.
- Sheu, B.C.; Hsu, S.M.; Ho, H.N.; Lien, H.C.; Huang, S.C.; Lin, R.H. A novel role of metalloproteinase in cancer-mediated immunosuppression. Cancer Res. 2001, 61, 237–242.
- Guedez, L.; Jensen-Taubman, S.; Bourboulia, D.; Kwityn, C.J.; Wei, B.; Caterina, J.; Stetler-Stevenson, W.G. TIMP-2 targets tumor-associated myeloid suppressor cells with effects in cancer immune dysfunction and angiogenesis. J. Immunother. 2012, 35, 502–512.
- Shay, G.; Lynch, C.C.; Fingleton, B. Moving targets: Emerging roles for MMPs in Cancer Progression and Metastasis. Matrix Biol. 2015, 44, 200–206.
- Rucci, N.; Sanità, P.; Angelucci, A. Roles of metalloproteases in metastatic niche. Curr. Mol. Med. 2011, 11, 609–622.
- Voura, E.B.; English, J.L.; Yu, H.-Y.E.; Ho, A.T.; Subarsky, P.; Hill, R.P.; Hojilla, C.V.; Khokha, R. Proteolysis during Tumor Cell Extravasation In Vitro: Metalloproteinase Involvement across Tumor Cell Types. PLoS ONE 2013, 8, e78413.
- Strand, S.; Vollmer, P.; van den Abeelen, L.; Gottfried, D.; Alla, V.; Heid, H.; Kuball, J.; Theobald, M.; Galle, P.R.; Strand, D. Cleavage of CD95 by matrix metalloproteinase-7 induces apoptosis resistance in tumour cells. Oncogene 2004, 23, 3732–3736.
- Newby, A.C. Matrix metalloproteinases regulate migration, proliferation, and death of vascular smooth muscle cells by degrading matrix and non-matrix substrates. Cardiovasc. Res. 2006, 69, 614–624.
- George, S.J.; Dwivedi, A. MMPs, cadherins, and cell proliferation. Trends Cardiovasc. Med. 2004, 14, 100–105.
- Kamiyama, M.; Naguro, I.; Ichijo, H. In vivo gene manipulation reveals the impact of stress-responsive MAPK pathways on tumor progression. Cancer Sci. 2015, 106, 785–796.
- Wortzel, I.; Seger, R. The ERK Cascade. Genes Cancer 2011, 2, 195–209.
- Kim, E.K.; Choi, E.-J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882.
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83.
- Olea-Flores, M.; Zuñiga-Eulogio, M.D.; Mendoza-Catalán, M.A.; Rodríguez-Ruiz, H.A.; Castañeda-Saucedo, E.; Ortuño-Pineda, C.; Padilla-Benavides, T.; Navarro-Tito, N. Extracellular-Signal Regulated Kinase: A Central Molecule Driving Epithelial–Mesenchymal Transition in Cancer. Int. J. Mol. Sci. 2019, 20, 2885.
- Yang, M.; Huang, C.-Z. Mitogen-activated protein kinase signaling pathway and invasion and metastasis of gastric cancer. World J. Gastroenterol. 2015, 21, 11673–11679.
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102.
- Fiore, M.; Forli, S.; Manetti, F. Targeting Mitogen-activated Protein Kinase-activated Protein Kinase 2 (MAPKAPK2, MK2): Medicinal Chemistry Efforts to Lead Small Molecule Inhibitors to Clinical Trials. J. Med. Chem. 2016, 59, 3609–3634.
- Kiniwa, Y.; Okuyama, R. Recent advances in molecular targeted therapy for unresectable and metastatic BRAF-mutated melanoma. Jpn. J. Clin. Oncol. 2021, 51, 315–320.
- Djanani, A.; Eller, S.; Öfner, D.; Troppmair, J.; Maglione, M. The Role of BRAF in Metastatic Colorectal Carcinoma–Past, Present, and Future. Int. J. Mol. Sci. 2020, 21, 9001.
- Proietti, I.; Skroza, N.; Bernardini, N.; Tolino, E.; Balduzzi, V.; Marchesiello, A.; Michelini, S.; Volpe, S.; Mambrin, A.; Mangino, G.; et al. Mechanisms of Acquired BRAF Inhibitor Resistance in Melanoma: A Systematic Review. Cancers 2020, 12, E2801.
- Guo, Y.-J.; Pan, W.-W.; Liu, S.-B.; Shen, Z.-F.; Xu, Y.; Hu, L.-L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007.
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, E1618.
- Gao, J.; Wang, Y.; Yang, J.; Zhang, W.; Meng, K.; Sun, Y.; Li, Y.; He, Q.-Y. RNF128 Promotes Invasion and Metastasis Via the EGFR/MAPK/MMP-2 Pathway in Esophageal Squamous Cell Carcinoma. Cancers 2019, 11, E840.
- Chang, M.-C.; Chen, C.-A.; Chen, P.-J.; Chiang, Y.-C.; Chen, Y.-L.; Mao, T.-L.; Lin, H.-W.; Lin Chiang, W.-H.; Cheng, W.-F. Mesothelin enhances invasion of ovarian cancer by inducing MMP-7 through MAPK/ERK and JNK pathways. Biochem. J. 2012, 442, 293–302.
- Lu, L.; Wang, J.; Wu, Y.; Wan, P.; Yang, G. Rap1A promotes ovarian cancer metastasis via activation of ERK/p38 and notch signaling. Cancer Med. 2016, 5, 3544–3554.
- Glading, A.; Chang, P.; Lauffenburger, D.A.; Wells, A. Epidermal growth factor receptor activation of calpain is required for fibroblast motility and occurs via an ERK/MAP kinase signaling pathway. J. Biol. Chem. 2000, 275, 2390–2398.
- Glading, A.; Uberall, F.; Keyse, S.M.; Lauffenburger, D.A.; Wells, A. Membrane proximal ERK signaling is required for M-calpain activation downstream of epidermal growth factor receptor signaling. J. Biol. Chem. 2001, 276, 23341–23348.
- Glading, A.; Lauffenburger, D.A.; Wells, A. Cutting to the chase: Calpain proteases in cell motility. Trends Cell Biol. 2002, 12, 46–54.
- Glading, A.; Bodnar, R.J.; Reynolds, I.J.; Shiraha, H.; Satish, L.; Potter, D.A.; Blair, H.C.; Wells, A. Epidermal growth factor activates m-calpain (calpain II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol. Cell. Biol. 2004, 24, 2499–2512.
- Chen, P.; Xie, H.; Sekar, M.C.; Gupta, K.; Wells, A. Epidermal growth factor receptor-mediated cell motility: Phospholipase C activity is required, but mitogen-activated protein kinase activity is not sufficient for induced cell movement. J. Cell Biol. 1994, 127, 847–857.
- Chen, P.; Murphy-Ullrich, J.E.; Wells, A. A role for gelsolin in actuating epidermal growth factor receptor-mediated cell motility. J. Cell Biol. 1996, 134, 689–698.
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389.
- Pinto-Díez, C.; Ferreras-Martín, R.; Carrión-Marchante, R.; González, V.M.; Martín, M.E. Deeping in the Role of the MAP-Kinases Interacting Kinases (MNKs) in Cancer. Int. J. Mol. Sci. 2020, 21, 2967.
- Jiang, J.; Wang, K.; Chen, Y.; Chen, H.; Nice, E.C.; Huang, C. Redox regulation in tumor cell epithelial-mesenchymal transition: Molecular basis and therapeutic strategy. Signal Transduct. Target. Ther. 2017, 2, 17036.
- Wang, Z.; Li, Y.; Sarkar, F.H. Signaling Mechanism(S) of Reactive Oxygen Species in Epithelial-Mesenchymal Transition Reminiscent of Cancer Stem Cells in Tumor Progression. Curr. Stem Cell Res. Ther. 2010, 5, 74–80.
- Lee, S.Y.; Ju, M.K.; Jeon, H.M.; Lee, Y.J.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Reactive oxygen species induce epithelial-mesenchymal transition, glycolytic switch, and mitochondrial repression through the Dlx-2/Snail signaling pathways in MCF-7 cells. Mol. Med. Rep. 2019, 20, 2339–2346.
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735.
- Chen, C.; Nelson, L.J.; Ávila, M.A.; Cubero, F.J. Mitogen-Activated Protein Kinases (MAPKs) and Cholangiocarcinoma: The Missing Link. Cells 2019, 8, 1172.
- Wu, X.; Zhang, W.; Font-Burgada, J.; Palmer, T.; Hamil, A.S.; Biswas, S.K.; Poidinger, M.; Borcherding, N.; Xie, Q.; Ellies, L.G.; et al. Ubiquitin-conjugating enzyme Ubc13 controls breast cancer metastasis through a TAK1-p38 MAP kinase cascade. Proc. Natl. Acad. Sci. USA 2014, 111, 13870–13875.
- del Barco Barrantes, I.; Nebreda, A.R. Roles of p38 MAPKs in invasion and metastasis. Biochem. Soc. Trans. 2012, 40, 79–84.
- Zou, X.; Blank, M. Targeting p38 MAP kinase signaling in cancer through post-translational modifications. Cancer Lett. 2017, 384, 19–26.
- Whitmarsh, A.J. A central role for p38 MAPK in the early transcriptional response to stress. BMC Biol. 2010, 8, 47.
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417.
- Trempolec, N.; Dave-Coll, N.; Nebreda, A.R. SnapShot: p38 MAPK substrates. Cell 2013, 152, 924.
- Hong, J.; Zhou, J.; Fu, J.; He, T.; Qin, J.; Wang, L.; Liao, L.; Xu, J. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011, 71, 3980–3990.
- Ansieau, S.; Bastid, J.; Doreau, A.; Morel, A.-P.; Bouchet, B.P.; Thomas, C.; Fauvet, F.; Puisieux, I.; Doglioni, C.; Piccinin, S.; et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 2008, 14, 79–89.
- Weiss, M.B.; Abel, E.V.; Mayberry, M.M.; Basile, K.J.; Berger, A.C.; Aplin, A.E. TWIST1 is an ERK1/2 effector that promotes invasion and regulates MMP-1 expression in human melanoma cells. Cancer Res. 2012, 72, 6382–6392.
- Hsieh, M.-J.; Chen, K.-S.; Chiou, H.-L.; Hsieh, Y.-S. Carbonic anhydrase XII promotes invasion and migration ability of MDA-MB-231 breast cancer cells through the p38 MAPK signaling pathway. Eur. J. Cell Biol. 2010, 89, 598–606.
- Johansson, N.; Ala-aho, R.; Uitto, V.; Grénman, R.; Fusenig, N.E.; López-Otín, C.; Kähäri, V.M. Expression of collagenase-3 (MMP-13) and collagenase-1 (MMP-1) by transformed keratinocytes is dependent on the activity of p38 mitogen-activated protein kinase. J. Cell Sci. 2000, 113, 227–235.
- Kumar, P.; Yadav, A.; Patel, S.N.; Islam, M.; Pan, Q.; Merajver, S.D.; Teknos, T.N. Tetrathiomolybdate inhibits head and neck cancer metastasis by decreasing tumor cell motility, invasiveness and by promoting tumor cell anoikis. Mol. Cancer 2010, 9, 206.
- Park, S.Y.; Jeong, K.J.; Panupinthu, N.; Yu, S.; Lee, J.; Han, J.W.; Kim, J.M.; Lee, J.-S.; Kang, J.; Park, C.G.; et al. Lysophosphatidic acid augments human hepatocellular carcinoma cell invasion through LPA1 receptor and MMP-9 expression. Oncogene 2011, 30, 1351–1359.
- Xu, L.; Chen, S.; Bergan, R.C. MAPKAPK2 and HSP27 are downstream effectors of p38 MAP kinase-mediated matrix metalloproteinase type 2 activation and cell invasion in human prostate cancer. Oncogene 2006, 25, 2987–2998.
- Hipp, S.; Berg, D.; Ergin, B.; Schuster, T.; Hapfelmeier, A.; Walch, A.; Avril, S.; Schmalfeldt, B.; Höfler, H.; Becker, K.-F. Interaction of Snail and p38 mitogen-activated protein kinase results in shorter overall survival of ovarian cancer patients. Virchows Arch. 2010, 457, 705–713.
- Ricciardi, M.; Zanotto, M.; Malpeli, G.; Bassi, G.; Perbellini, O.; Chilosi, M.; Bifari, F.; Krampera, M. Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. Br. J. Cancer 2015, 112, 1067–1075.
- Zhou, J.; Zhang, C.; Pan, J.; Chen, L.; Qi, S.-T. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human adamantinomatous craniopharyngioma cells and promotes tumor cell migration. Mol. Med. Rep. 2017, 15, 4123–4131.
- Sun, Q.; Shang, Y.; Sun, F.; Dong, X.; Niu, J.; Li, F. Interleukin-6 Promotes Epithelial-Mesenchymal Transition and Cell Invasion through Integrin β6 Upregulation in Colorectal Cancer. Oxid. Med. Cell. Longev. 2020, 2020, 8032187.
- Lyons, J.G.; Patel, V.; Roue, N.C.; Fok, S.Y.; Soon, L.L.; Halliday, G.M.; Gutkind, J.S. Snail up-regulates pro-inflammatory mediators and inhibits differentiation in oral keratinocytes. Cancer Res. 2008, 68, 4525–4530.
- Huang, Q.; Lan, F.; Wang, X.; Yu, Y.; Ouyang, X.; Zheng, F.; Han, J.; Lin, Y.; Xie, Y.; Xie, F.; et al. IL-1β-induced activation of p38 promotes metastasis in gastric adenocarcinoma via upregulation of AP-1/c-fos, MMP2 and MMP9. Mol. Cancer 2014, 13, 18.
- Chen, X.; Wei, H.; Li, J.; Liang, X.; Dai, S.; Jiang, L.; Guo, M.; Qu, L.; Chen, Z.; Chen, L.; et al. Structural basis for DNA recognition by FOXC2. Nucleic Acids Res. 2019, 47, 3752–3764.
- Zhang, Y.; Liao, Y.; Chen, C.; Sun, W.; Sun, X.; Liu, Y.; Xu, E.; Lai, M.; Zhang, H. p38-regulated FOXC1 stability is required for colorectal cancer metastasis. J. Pathol. 2020, 250, 217–230.
- Werden, S.J.; Sphyris, N.; Sarkar, T.R.; Paranjape, A.N.; LaBaff, A.M.; Taube, J.H.; Hollier, B.G.; Ramirez-Peña, E.Q.; Soundararajan, R.; den Hollander, P.; et al. Phosphorylation of serine 367 of FOXC2 by p38 regulates ZEB1 and breast cancer metastasis, without impacting primary tumor growth. Oncogene 2016, 35, 5977–5988.
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487.
- Emerling, B.M.; Platanias, L.C.; Black, E.; Nebreda, A.R.; Davis, R.J.; Chandel, N.S. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol. Cell. Biol. 2005, 25, 4853–4862.
- López-Novoa, J.M.; Nieto, M.A. Inflammation and EMT: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009, 1, 303–314.
- Laferriere, J.; Houle, F.; Taher, M.M.; Valerie, K.; Huot, J. Transendothelial migration of colon carcinoma cells requires expression of E-selectin by endothelial cells and activation of stress-activated protein kinase-2 (SAPK2/p38) in the tumor cells. J. Biol. Chem. 2001, 276, 33762–33772.
- Kobayashi, M.; Nishita, M.; Mishima, T.; Ohashi, K.; Mizuno, K. MAPKAPK-2-mediated LIM-kinase activation is critical for VEGF-induced actin remodeling and cell migration. EMBO J. 2006, 25, 713–726.
- Wang, W.; Eddy, R.; Condeelis, J. The cofilin pathway in breast cancer invasion and metastasis. Nat. Rev. Cancer 2007, 7, 429–440.
- Naffa, R.; Vogel, L.; Hegedűs, L.; Pászty, K.; Tóth, S.; Kelemen, K.; Singh, N.; Reményi, A.; Kállay, E.; Cserepes, M.; et al. P38 MAPK Promotes Migration and Metastatic Activity of BRAF Mutant Melanoma Cells by Inducing Degradation of PMCA4b. Cells 2020, 9, E1209.
- Wada, M.; Canals, D.; Adada, M.; Coant, N.; Salama, M.F.; Helke, K.L.; Arthur, J.S.; Shroyer, K.R.; Kitatani, K.; Obeid, L.M.; et al. P38 delta MAPK promotes breast cancer progression and lung metastasis by enhancing cell proliferation and cell detachment. Oncogene 2017, 36, 6649–6657.
- Xu, M.; Wang, S.; Wang, Y.; Wu, H.; Frank, J.A.; Zhang, Z.; Luo, J. Role of p38γ MAPK in regulation of EMT and cancer stem cells. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3605–3617.
- Fang, Y.; Wang, J.; Wang, G.; Zhou, C.; Wang, P.; Zhao, S.; Zhao, S.; Huang, S.; Su, W.; Jiang, P.; et al. Inactivation of p38 MAPK contributes to stem cell-like properties of non-small cell lung cancer. Oncotarget 2017, 8, 26702–26717.
- Ebelt, N.D.; Cantrell, M.A.; Van Den Berg, C.L. c-Jun N-Terminal Kinases Mediate a Wide Range of Targets in the Metastatic Cascade. Genes Cancer 2013, 4, 378–387.
- Li, Y.-S.; Deng, Z.-H.; Zeng, C.; Lei, G.-H. JNK pathway in osteosarcoma: Pathogenesis and therapeutics. J. Recept. Signal Transduct. Res. 2016, 36, 465–470.
- Tam, S.Y.; Law, H.K.-W. JNK in Tumor Microenvironment: Present Findings and Challenges in Clinical Translation. Cancers 2021, 13, 2196.
- Tournier, C. The 2 Faces of JNK Signaling in Cancer. Genes Cancer 2013, 4, 397–400.
- Sabapathy, K.; Hochedlinger, K.; Nam, S.Y.; Bauer, A.; Karin, M.; Wagner, E.F. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol. Cell 2004, 15, 713–725.
- Shaulian, E.; Karin, M. AP-1 in cell proliferation and survival. Oncogene 2001, 20, 2390–2400.
- Cellurale, C.; Sabio, G.; Kennedy, N.J.; Das, M.; Barlow, M.; Sandy, P.; Jacks, T.; Davis, R.J. Requirement of c-Jun NH(2)-terminal kinase for Ras-initiated tumor formation. Mol. Cell. Biol. 2011, 31, 1565–1576.
- Hammouda, M.B.; Ford, A.E.; Liu, Y.; Zhang, J.Y. The JNK Signaling Pathway in Inflammatory Skin Disorders and Cancer. Cells 2020, 9, E857.
- Guma, M.; Firestein, G.S. c-Jun N-Terminal Kinase in Inflammation and Rheumatic Diseases. Open Rheumatol. J. 2012, 6, 220–231.
- Kaminska, B. Molecular characterization of inflammation-induced JNK/c-Jun signaling pathway in connection with tumorigenesis. Methods Mol. Biol. 2009, 512, 249–264.
- Takahashi, H.; Ogata, H.; Nishigaki, R.; Broide, D.H.; Karin, M. Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta- and JNK1-dependent inflammation. Cancer Cell 2010, 17, 89–97.
- Krejsgaard, T.; Vetter-Kauczok, C.S.; Woetmann, A.; Lovato, P.; Labuda, T.; Eriksen, K.W.; Zhang, Q.; Becker, J.C.; Ødum, N. Jak3- and JNK-dependent vascular endothelial growth factor expression in cutaneous T-cell lymphoma. Leukemia 2006, 20, 1759–1766.
- Saníger, M.L.; Oya, R.; Macías, D.; Domínguez, J.N.; Aránega, A.; Luque, F. c-Jun kinase mediates expression of VEGF induced at transcriptional level by Rac1 and Cdc42Hs but not by RhoA. J. Cell. Biochem. 2006, 98, 650–660.
- Igaki, T.; Pagliarini, R.A.; Xu, T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr. Biol. 2006, 16, 1139–1146.
- Wei, P.-L.; Huang, C.-Y.; Tai, C.-J.; Batzorig, U.; Cheng, W.-L.; Hunag, M.-T.; Chang, Y.-J. Glucose-regulated protein 94 mediates metastasis by CCT8 and the JNK pathway in hepatocellular carcinoma. Tumour Biol. 2016, 37, 8219–8227.
- Yang, M.; Gu, Y.-Y.; Peng, H.; Zhao, M.; Wang, J.; Huang, S.-K.; Yuan, X.-H.; Li, J.; Sang, J.-L.; Luo, Q.; et al. NAIF1 inhibits gastric cancer cells migration and invasion via the MAPK pathways. J. Cancer Res. Clin. Oncol. 2015, 141, 1037–1047.
- Lv, B.; Shi, T.; Wang, X.; Song, Q.; Zhang, Y.; Shen, Y.; Ma, D.; Lou, Y. Overexpression of the novel human gene, nuclear apoptosis-inducing factor 1, induces apoptosis. Int. J. Biochem. Cell Biol. 2006, 38, 671–683.
- Zhou, H.-M.; Fang, Y.-Y.; Weinberger, P.M.; Ding, L.-L.; Cowell, J.K.; Hudson, F.Z.; Ren, M.; Lee, J.R.; Chen, Q.-K.; Su, H.; et al. Transgelin increases metastatic potential of colorectal cancer cells in vivo and alters expression of genes involved in cell motility. BMC Cancer 2016, 16, 55.
- Lin, Y.; Buckhaults, P.J.; Lee, J.R.; Xiong, H.; Farrell, C.; Podolsky, R.H.; Schade, R.R.; Dynan, W.S. Association of the actin-binding protein transgelin with lymph node metastasis in human colorectal cancer. Neoplasia 2009, 11, 864–873.
- Zhou, H.; Zhang, Y.; Chen, Q.; Lin, Y. AKT and JNK Signaling Pathways Increase the Metastatic Potential of Colorectal Cancer Cells by Altering Transgelin Expression. Dig. Dis. Sci. 2016, 61, 1091–1097.
- Cai, J.; Du, S.; Wang, H.; Xin, B.; Wang, J.; Shen, W.; Wei, W.; Guo, Z.; Shen, X. Tenascin-C induces migration and invasion through JNK/c-Jun signalling in pancreatic cancer. Oncotarget 2017, 8, 74406–74422.
- Chen, P.-C.; Tang, C.-H.; Lin, L.-W.; Tsai, C.-H.; Chu, C.-Y.; Lin, T.-H.; Huang, Y.-L. Thrombospondin-2 promotes prostate cancer bone metastasis by the up-regulation of matrix metalloproteinase-2 through down-regulating miR-376c expression. J. Hematol. Oncol. 2017, 10, 33.
- Du, R.; Tang, G.; Tang, Z.; Kuang, Y. Ectopic expression of CC chemokine receptor 7 promotes prostate cancer cells metastasis via Notch1 signaling. J. Cell. Biochem. 2019, 120, 9639–9647.
- Swenson-Fields, K.I.; Sandquist, J.C.; Rossol-Allison, J.; Blat, I.C.; Wennerberg, K.; Burridge, K.; Means, A.R. MLK3 limits activated Galphaq signaling to Rho by binding to p63RhoGEF. Mol. Cell 2008, 32, 43–56.
- Chen, J.; Gallo, K.A. MLK3 regulates paxillin phosphorylation in chemokine-mediated breast cancer cell migration and invasion to drive metastasis. Cancer Res. 2012, 72, 4130–4140.
- Tsubouchi, A.; Sakakura, J.; Yagi, R.; Mazaki, Y.; Schaefer, E.; Yano, H.; Sabe, H. Localized suppression of RhoA activity by Tyr31/118-phosphorylated paxillin in cell adhesion and migration. J. Cell Biol. 2002, 159, 673–683.
- Swenson, K.I.; Winkler, K.E.; Means, A.R. A new identity for MLK3 as an NIMA-related, cell cycle-regulated kinase that is localized near centrosomes and influences microtubule organization. Mol. Biol. Cell 2003, 14, 156–172.
- Nguyen, D.X.; Bos, P.D.; Massagué, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284.
- Koury, J.; Zhong, L.; Hao, J. Targeting Signaling Pathways in Cancer Stem Cells for Cancer Treatment. Stem Cells Int. 2017, 2017, 2925869.
- Rattanasinchai, C.; Gallo, K.A. MLK3 Signaling in Cancer Invasion. Cancers 2016, 8, 51.