Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Cancer contains tumor-initiating stem-like cells (TICs) that are resistant to therapies. Experimental evidence indicates that hepatocellular carcinoma (HCC) and TIC development are influenced by permissive conditions in response to changes in gut microbiota.
- cancer stem cell (CSC)
- tumor-initiating stem-like cells (TICs)
- hepatocellular carcinoma (HCC)
1. Introduction
Treatment options for HCC are limited. The 3- or 5-year survival rate of HCC is 13–21% and 5%, respectively, without any curative treatment in advanced countries such as the U.S. [1][2][3][4][5]. The incidence rate of extrahepatic metastasis is 13% at 5 years [6][7]. Currently liver resection is the only viable option for HCC combined with cirrhosis that is the terminal stage of fibrosis, leading to hyperplasia formation [8]. Currently, only 10–23% of HCC patients are candidates for surgery [9][10][11]. Thus, HCV-associated HCC remains an incurable malignancy and an urgent unmet medical need. As 40% of HCCs are derived from TICs, TIC-mediated HCC development must be considered as clinically important.
TICs are resistant to conventional chemotherapy and immunotherapy and persist as recurrent tumors or circulating tumor cells (CTC) [12]. TICs express a core pluripotency-associated transcription factor (TF) network [13][14]. Up to forty percent of HCCs have clonality and are considered to originate from progenitor/stem cells [15][16][17][18]. TICs express stemness genes that are also expressed in pluripotent stem cells, including CD133 (Prominin in mice), Wnt/β-catenin, Nanog [19], NOTCH, Hedgehog/SMO, BMI, OCT3/4 [20][21][22][23][24][25][26][27][28][29][30][31], CD44 (cell adhesion molecule), and CD34. CD133+/CD49f+ HCC TICs confer resistance to chemotherapy, which hampers efficacy of therapy in HCC [32]. TICs exhibit a loss of this intrinsic asymmetry, leading to subsequent unchecked expansion of the progenitor cell pool [33][34][35][36][37][38]. Cell-fate-determinant molecule NUMB, and p53-MDM2-associated proteins, are targeted by interacting protein TBC1D15 in TICs [39]. These stemness factors are commonly expressed in TICs and pluripotent stem cells. Stemness factors promote therapy resistance and self-renewal ability.
1.1. Challenge in Targeting of Actionable Mutations
There are no current targeted therapy options for the most prevalent mutations (most are not “actionable”). HCC has only 2.5% of actionable mutations that can be clinically targeted by FDA-approved drugs, while biliary cancer has 45% actionable mutations based on Oncokb.org (Level 3A, Level 3B) and HCC tumor genetics in a TCGA cohort [40]. These indicate many HCC mutations do not have conventional therapeutic targets. Therefore, the role of immunotherapy for the treatment of these diseases is an area of intense investigation [40].
1.2. Molecular Tumor Board (MTB) Review and Actionable Mutations in Liver Cancer
The molecular tumor board (MTB) review can guide choices of therapy for actionable mutations, clarify diagnosis, and identify patients who require germline testing. Prospective clinical sequencing of 10,000 patients revealed the mutational landscape of metastatic cancer [41] Clinical actionability of somatic alterations revealed by MSK-IMPACT was the lowest in HCC mutations at 2.5% [41][42][43][44][45], while clinical actionability of somatic alterations revealed by MSK-IMPACT showed that 45% of biliary cancer mutations are clinically actionable. The molecular tumor board (MTB) for intrahepatic cholangiocarcinoma (iCCA) shows clinically targetable mutations [46][47][48]. iCCA is a heterogeneous disease with several identifiable genetic driver mutations (i.e., FGFR2-fusions IDH mutations, etc.) [40]. For iCCA, fluorescence in situ hybridization (FISH), DNA/RNA-seq, and immunohistochemistry (IHC) analyses can identify cancer-driver mutations, including IDH1/2, CDK4/67, PRKACA/B, and BRCA1/2. FGFR2 and NYRK fusions, BRAF and IDH1 mutations, and microsatellite instability high (MSI-H)/dMMR (defective mismatch DNA repair) predict responses to targeted/immune therapies [41]. Tumor next-generation sequencing (NGS) should be considered in selected HCC patients with atypical histology/diagnostic features or who may be eligible for clinical trials. HCC classification, cells of origin, genetic and epigenetic abnormalities, molecular alterations, biomarker discovery, and treatments of iCCA have been well characterized [49].
2. Gut-Microbiota-Mediated Immune Regulatory Mechanisms by Immunotherapy
2.1. Bacterial Enzyme Inhibitors Can Be Used for Treatment
Gut microbial produced metabolites can be recognized by host pathogen recognition sensors to promote HCC progression. Metabolism of dietary components by the gut microbiota produces short-chain fatty acids, including other metabolites. When combined with microorganism fragments, these can stimulate the meta-organismal endocrine axis to promote HCC onset and growth. For example, trimethylamine (TMA) produced in the gut promotes ALD [44]. Thus, pharmacological interventions at the level of the gut microbiome should reduce HCC risk.
Targeting of the gut microbiota has great potential as a therapeutic modality for many diseases. However, relatively little is known regarding the contribution of commensal bacteria to normal host physiological functions [45]. For example, it was reported that 11 bacterial strains in feces obtained from normal human donors induce CD8 T cells to produce IFN-γ in the intestine in the absence of a generalized inflammatory response dependent on CD103+ DC and MHC class Ia [45]. These 11 strains also improved the efficacy of immune checkpoint inhibitors and aided host suppression against Listeria monocytogenes infection [45]. Thus, these 11 identified strains, which represent low-abundance components of the human microbiome, are potential biotherapeutics [45].
2.2. TLR2 Signaling in DCs Promotes Treg Differentiation to Attenuate the Inflammation
TLR2 senses components from bacteria, mycoplasma, fungi, and viruses [47] to activate NF-κB to promote a Th17 cell response to enhance the inflammation response and anti-inflammation responses [48][50]. Lactobacillus acidophilus stimulates the TLR2 pathway of murine myeloid dendritic cells (mDC) to induce interferon-β (IFN-β), while IL-10 secretion in plasmacytoid DC (pDC) is TLR9 dependent (Figure 1). Bifidobacterium infantis 35624 stimulates the TLR2/TLR6 pathway to increase IL-10 secretion from human DCs. Polysaccharide A of Gram negative bacilli can activate TLR2 and promote the secretion of anti-inflammatory cytokine IL-10 [51]. These diverse immune responses depend on the appropriate co-receptor and microenvironment [48].
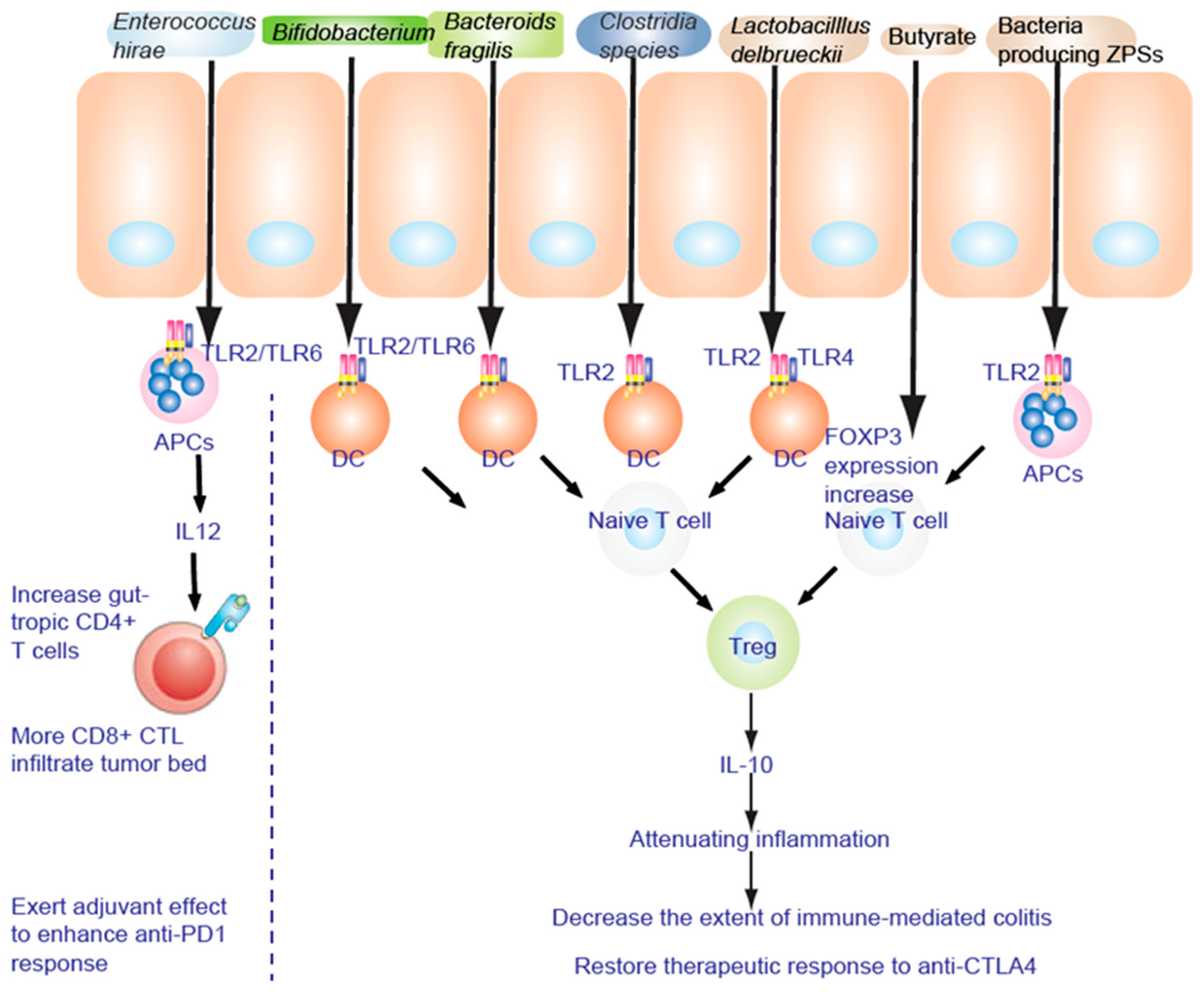
Figure 1. TLR2 is necessary to alleviate the inflammatory response. Fructo-oligosaccharide and inulin are considered as prebiotics, affecting IECs to be hyporesponsive to activation of NF-κB and MAPK induced by pathogens. NF-κB and MAPK reduce the inflammatory response to lipopolysaccharide (LPS).
2.3. Regulatory T Cells
Treg cells secrete the anti-inflammatory cytokine IL-10 to attenuate inflammation. IL-6, IL-21, and IL-2 dynamically regulate the balance between Th17 and Treg cell differentiation [52][53]. Intestinal bacteria act to stimulate and shape the T cell subsets. Short-chain fatty acid prime and induce Th17 cells undergo differentiation locally in the lamina propria. In addition, segmented filamentous bacteria antigen (SFB) adhesion to enterocytes stimulates serum amyloid A and ROS to induce Th17 cells [54]. MHCII-dependent antigen presentation of SFB occurs on DC [55] (Figure 1). Commensal bacteria (such as the Lachnospiraceae family, A4 bacteria) induce transforming growth factor β (TGF-β) production to inhibit Th2 cell development [56]. Clostridia colonization effects on T cell differentiation induce Treg cell expansion to suppress inflammation in mice [57][58]. In germ-free (GF) mice, colonization of gut bacteria and LPS-rich sterile diet induced T and B cell proliferation and differentiation in Peyer’s patches (PP) and mesenteric lymph nodes (MLN), especially by CD4+ Foxp3+ T cells in MLN [59]. Polysaccharides do affect T cell differentiation. To reinforce its intestinal colonization, polysaccharide A (PSA) from Bacteroides fragilis promotes Treg cell secretion and suppresses Th17 activity [60]. The growth of bacteria encoding zwitterionic capsular polysaccharides (ZPS), as shown by genomic screen, results in stimulation of T cell differentiation of Treg cells and IL-10 production mediated by antigen presenting cells (APC) [61].
Zwitterionic polysaccharides bind the TLR2 complex on CD11b+ DC to mobilize lamina propria CD11b+ DC. This in turn stimulates Treg differentiation to promote anergy against immunity induced by CTLA-4 blockade [62] via interleukin-12 (IL-12)-dependent cognate TH1 immune responses against Bf capsular polysaccharides (Figure 1). CTLA4-mediated TH1 immune response is blocked by Treg to protect against experimental abscess formation [62] independent of TLR2/TLR4-mediated innate signaling [63][64]. A clustering of genus composition of stools [65][66] distinguished three clusters with Alloprevotella or Prevotella driving cluster A and distinct Bacteroides spp. driving clusters B and C. During anti-CTLA4 (ipilimumab) therapy, the proportions of MM patients falling into cluster C increased at the expense of those belonging to cluster B through the colonization of the immunogenic bacteria Bf and Bt [62][63][64][67][68][69].
2.4. Commensal Bacteria-Derived Products Stimulate DCs and Regulate Tregs
High-alcohol-producing Klebsiella pneumoniae causes fatty liver disease [70]. Intestinal microbiota in human stool contributes to susceptibility to ALD shown by the use of ALD-FMT in germ-free mice [71][72]. To edit gut microbiota, four distinct bacteriophages (podophages of the virulent Picovirinae group) were isolated from sewage water. Feeding of four podophages of the virulent Picovirinae group lyse the cytolytic E. faecalis strain [73]. Gavage of bacteriophages that target cytolytic E. faecalis attenuates alcoholic liver disease that promotes E. faecalis expansion (2700-fold increase) by reducing steatosis, inflammation, and liver injury of mice chronically fed ethanol [74]. Therefore, the gut microbiome is a potential therapeutic target in the pathogenesis (pro-inflammatory response) and treatment of chronic liver disease [75], since it is altered in liver cirrhosis [76]. Overgrowth by Clostridiales, Streptococcus, Lactobacillus, Bacteroides, and Enterobacteriaceae genera promotes gut injury and liver disease. In liver cirrhosis, Bacteroides increase while Firmicutes decrease. Rifaximin inhibits oral-originating species and selectively decontaminates the gut. Further environmental factors mediating microbiota changes can promote excessive inflammatory signaling.
2.5. A Live Microbiome Co-Culture in a Gut-on-a-Chip Microfluidic Device
A live microbiome was co-cultured with micro-engineered human intestinal villi in a gut-on-a-chip microfluidic device [77]. The intestine–liver axis on-chip reveals the intestinal protective role on hepatic damage (Figure 1) by emulating ethanol first-pass metabolism [78][79][80][81]. Those who live longer customarily consume the following foods, including pasta (barley: fibers), soybean (flavone), seaweed (mineral), seafood (fish oil: DHA, BHA), and green tea (polyphenols, catechin epigallocatechin-3-gallate: EGCG). Prebiotics are nondigestible dietary supplements, including mucin or long-chain carbohydrates, which promote proliferation of beneficial commensal bacteria and improve the ecological balance of the gut. The effects of prebiotics can be tested in this system (Figure 1).
Synbiotic treatment normalizes gut microbiota and concomitantly reduces toxic gut microbiota to repair leaky gut syndrome [82]. These bacteria digest prebiotics to produce short-chain fatty acids which inhibit intestinal pathogen growth, provide enterocyte nutrition (butyrate), and promote mineral absorption. Bifidobacterium growth is enhanced with a prebiotic-containing formula (90% short-chain galacto-oligosaccharide, 10% long-chain fructo-oligosaccharide), fructo-oligosaccharides [83], and inulin [84].
Patients who responded to nivolumab (PD-1 antibody) were enriched with Bacteroides caccae [85] and Fecalibacterium prausnitzii, Bacteroides thetaiotamicron, and Holdemania filiformis, whereas patients who responded to pembrolizumab (another PD-1 antibody) showed that their gut microbiota was enriched with Dorea formicogenerans. This treatment increased bacterial diversity and abundance of bacteria from Akkermansia muciniphila [86], Bifidobacterium spp. (B. longum, Collinsella aerofaciens) [87], Enterococcus faecium, and the Ruminococcaceae family, to induce Treg accumulation by cooperating with DC in the colon (Figure 1).
2.6. TLR2 Is Necessary to Alleviate the Inflammatory Response
TLR2 senses components from bacteria, mycoplasma, fungi, and viruses [47]. TLR2 signaling induces both pro- and anti-inflammatory responses. Bifidobacterium infantis 35624 treatment increases IL-10 secretion through the TLR2/TLR6 pathway in human myeloid dendritic cell (mDC) and monocyte-derived DC (MDDC), while IL-10 secretion in plasmacytoid DC (pDC) is TLR9 dependent. Tregs secrete the anti-inflammatory cytokine IL-10 to attenuate inflammation (Figure 1). Therefore, feeding of FMD + synbiotics preconditions gut microbiota and repairs the leaky gut to improve immunotherapy and chemotherapy (Figure 1).
2.7. Metabolism and Local Effects of SCFAs
Fermentation of dietary fiber in the colon generates short-chain (ranging from one to six carbon atoms) saturated fatty acids (SCFAs) [88]. Production of SCFA is dependent on dietary fiber and can result in gut production of approximately 500–600 mmol of SCFAs per day [89]. Acetate (C2) is the most abundant SCFA in the human body, followed by propionate (C3) and butyrate (C4) (in a molar ratio of 60:20:20, dependent on microbiota composition) as the most abundant anions in the colon [90][91]. Bowel movements transfer gut contents from the terminal ileum to the proximal colon where SCFAs can reduce the pH. Lesser amounts of other SCFAs, including caproate, formate, and valerate, are also produced [90]. Monocarboxylate transporters (MCTs) allow SCFA absorption by colonocytes in a H+-dependent, electroneutral manner, whereas the electrogenic, sodium-dependent monocarboxylate transporter 1 (SMCT1; known as SLC5A8) transports the SCFA anion [92].
Because of microbiota changes or intestinal microbiota transplantation in liver diseases and cirrhosis, use of pharmacotherapeutics must be cognizant of these issues when considering treatment options [93]. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome [94]. Allogenic fecal microbiota transplantation in patients with NAFLD improves abnormal small intestinal permeability, as shown in a randomized control trial [95]. Alkaline phosphatase can be used as a surrogate marker for liver–gut changes. C. difficile with cirrhosis is a deleterious combination with greater mortality via brain dysfunction due to SCFA downregulation. GF mice have altered microbial infection inflammatory markers (IL1β, MCP1, and IBA). Post-FMT GF mice recipients show improvement of neuro-inflammation [96]. Use of capsular fecal transplantation improves microbial function and supports better clinical outcomes in cirrhosis [97]. Oral capsule FMT (containing Ruminococcaceae) is currently under investigational new drug application (IND) guidance [98]. A randomized clinical trial of fecal microbiota transplant for alcohol use disorder is ongoing.
2.8. Exercise or Phage Therapy Retards Liver Diseases
Exercise reduces the incidence and progression of hepatocellular carcinoma in mouse models [99]. Personalized medicine approaches will stratify the HCC patient population into distinct subpopulations that may be responsive to HCC-type specific treatments [99]. As presented, there are several avenues of liver morbidities leading to HCC. For example, the microbiota is targeted for cytolysin+ alcoholic hepatitis patients. Future investigations will support a better understanding of antibiotic therapies for enteric pathogens, long-term effects of phage-based treatments, and use of precisely editing bacteria genomes by phage therapies (single phage or phage cocktail). These are all emerging areas of investigation, and these options reflect the original intent to reverse the triggering events leading to HCC.
2.9. Caveats for Fecal Microbiota Transplantation
FMT with multi-drug-resistant organisms (MDRO) can cause problems in donor recipient patients. Avoidance of C. difficile is important since it is responsible for the chronic liver diseases such as cirrhosis and alcoholic hepatitis. FMT trials for chronic liver diseases are currently in progress.
This entry is adapted from the peer-reviewed paper 10.3390/cancers14102381
References
- Barbara, L.; Benzi, G.; Gaiani, S.; Fusconi, F.; Zironi, G.; Siringo, S.; Rigamonti, A.; Barbara, C.; Grigioni, W.; Mazziotti, A.; et al. Natural history of small untreated hepatocellular carcinoma in cirrhosis: A multivariate analysis of prognostic factors of tumor growth rate and patient survival. Hepatology 1992, 16, 132–137.
- Ebara, M.; Ohto, M.; Shinagawa, T.; Sugiura, N.; Kimura, K.; Matsutani, S.; Morita, M.; Saisho, H.; Tsuchiya, Y.; Okuda, K. Natural history of minute hepatocellular carcinoma smaller than three centimeters complicating cirrhosis. A study in 22 patients. Gastroenterology 1986, 90, 289–298.
- El-Serag, H.B.; Mason, A.C. Rising incidence of hepatocellular carcinoma in the United States. N. Engl. J. Med. 1999, 340, 745–750.
- Liang, T.J.; Heller, T. Pathogenesis of hepatitis C-associated hepatocellular carcinoma. Gastroenterology 2004, 127, S62–S71.
- Okuda, K. Hepatocellular carcinoma. J. Hepatol. 2000, 32, 225–237.
- Kanda, M.; Tateishi, R.; Yoshida, H.; Sato, T.; Masuzaki, R.; Ohki, T.; Imamura, J.; Goto, T.; Yoshida, H.; Hamamura, K.; et al. Extrahepatic metastasis of hepatocellular carcinoma: Incidence and risk factors. Liver Int. 2008, 28, 1256–1263.
- Louis, P.; Scott, K.P.; Duncan, S.H.; Flint, H.J. Understanding the effects of diet on bacterial metabolism in the large intestine. J. Appl. Microbiol. 2007, 102, 1197–1208.
- Nakamura, Y.; Mizuguchi, T.; Tanimizu, N.; Ichinohe, N.; Ooe, H.; Kawamoto, M.; Meguro, M.; Hirata, K.; Mitaka, T. Preoperative hepatocyte transplantation improves the survival of rats with nonalcoholic steatohepatitis-related cirrhosis after partial hepatectomy. Cell Transplant. 2014, 23, 1243–1254.
- Shah, S.A.; Smith, J.K.; Li, Y.; Ng, S.C.; Carroll, J.E.; Tseng, J.F. Underutilization of therapy for hepatocellular carcinoma in the medicare population. Cancer 2011, 117, 1019–1026.
- Sonnenday, C.J.; Dimick, J.B.; Schulick, R.D.; Choti, M.A. Racial and geographic disparities in the utilization of surgical therapy for hepatocellular carcinoma. J. Gastrointest. Surg. 2007, 11, 1636–1646.
- McClain, C.J.; Barve, S.; Deaciuc, I.; Kugelmas, M.; Hill, D. Cytokines in alcoholic liver disease. Semin. Liver Dis. 1999, 19, 205–219.
- Massague, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306.
- Kim, J.; Woo, A.J.; Chu, J.; Snow, J.W.; Fujiwara, Y.; Kim, C.G.; Cantor, A.B.; Orkin, S.H. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 2010, 143, 313–324.
- Ikushima, H.; Todo, T.; Ino, Y.; Takahashi, M.; Saito, N.; Miyazawa, K.; Miyazono, K. Glioma-initiating cells retain their tumorigenicity through integration of the Sox axis and Oct4 protein. J. Biol. Chem. 2011, 286, 41434–41441.
- Alison, M.R. Liver stem cells: Implications for hepatocarcinogenesis. Stem. Cell Rev. 2005, 1, 253–260.
- Roskams, T. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene 2006, 25, 3818–3822.
- Zender, L.; Spector, M.S.; Xue, W.; Flemming, P.; Cordon-Cardo, C.; Silke, J.; Fan, S.T.; Luk, J.M.; Wigler, M.; Hannon, G.J.; et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006, 125, 1253–1267.
- Tang, Y.; Kitisin, K.; Jogunoori, W.; Li, C.; Deng, C.X.; Mueller, S.C.; Ressom, H.W.; Rashid, A.; He, A.R.; Mendelson, J.S.; et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 2445–2450.
- Feldman, D.E.; Chen, C.; Punj, V.; Tsukamoto, H.; Machida, K. Pluripotency factor-mediated expression of the leptin receptor (OB-R) links obesity to oncogenesis through tumor-initiating stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 829–834.
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The role of short-chain fatty acids in microbiota-gut-brain communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 461–478.
- Valk-Lingbeek, M.E.; Bruggeman, S.W.; van Lohuizen, M. Stem cells and cancer; the polycomb connection. Cell 2004, 118, 409–418.
- Chambers, I.; Smith, A. Self-renewal of teratocarcinoma and embryonic stem cells. Oncogene 2004, 23, 7150–7160.
- Beachy, P.A.; Karhadkar, S.S.; Berman, D.M. Tissue repair and stem cell renewal in carcinogenesis. Nature 2004, 432, 324–331.
- Bajaj, J.S.; Hays, R.A. Manipulation of the Gut-Liver Axis Using Microbiome Restoration Therapy in Primary Sclerosing Cholangitis. Am. J. Gastroenterol. 2019, 114, 1027–1029.
- Bajaj, J.S.; Kassam, Z.; Fagan, A.; Gavis, E.A.; Liu, E.; Cox, I.J.; Kheradman, R.; Heuman, D.; Wang, J.; Gurry, T.; et al. Fecal microbiota transplant from a rational stool donor improves hepatic encephalopathy: A randomized clinical trial. Hepatology 2017, 66, 1727–1738.
- Cai, X.; Chiu, Y.H.; Chen, Z.J. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 2014, 54, 289–296.
- Gibb, E.A.; Warren, R.L.; Wilson, G.W.; Brown, S.D.; Robertson, G.A.; Morin, G.B.; Holt, R.A. Activation of an endogenous retrovirus-associated long non-coding RNA in human adenocarcinoma. Genome Med. 2015, 7, 22.
- Llorente, C.; Schnabl, B. The gut microbiota and liver disease. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 275–284.
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185.
- Rao, A.; Kosters, A.; Mells, J.E.; Zhang, W.; Setchell, K.D.; Amanso, A.M.; Wynn, G.M.; Xu, T.; Keller, B.T.; Yin, H.; et al. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet-fed mice. Sci. Transl. Med. 2016, 8, 357ra122.
- Tan, F.P.Y.; Beltranena, E.; Zijlstra, R.T. Resistant starch: Implications of dietary inclusion on gut health and growth in pigs: A review. J. Anim. Sci. Biotechnol. 2021, 12, 124.
- Rountree, C.B.; Senadheera, S.; Mato, J.M.; Crooks, G.M.; Lu, S.C. Expansion of liver cancer stem cells during aging in methionine adenosyltransferase 1A-deficient mice. Hepatology 2008, 47, 1288–1297.
- Cicalese, A.; Bonizzi, G.; Pasi, C.E.; Faretta, M.; Ronzoni, S.; Giulini, B.; Brisken, C.; Minucci, S.; Di Fiore, P.P.; Pelicci, P.G. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell 2009, 138, 1083–1095.
- Knoblich, J.A. Asymmetric cell division: Recent developments and their implications for tumour biology. Nat. Rev. Mol. Cell Biol. 2010, 11, 849–860.
- Martin-Belmonte, F.; Perez-Moreno, M. Epithelial cell polarity, stem cells and cancer. Nat. Rev. Cancer 2012, 12, 23–38.
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340.
- Jiang, L.; Lang, S.; Duan, Y.; Zhang, X.; Gao, B.; Chopyk, J.; Schwanemann, L.K.; Ventura-Cots, M.; Bataller, R.; Bosques-Padilla, F.; et al. Intestinal Virome in Patients With Alcoholic Hepatitis. Hepatology 2020, 72, 2182–2196.
- Lang, S.; Demir, M.; Martin, A.; Jiang, L.; Zhang, X.; Duan, Y.; Gao, B.; Wisplinghoff, H.; Kasper, P.; Roderburg, C.; et al. Intestinal Virome Signature Associated With Severity of Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 159, 1839–1852.
- Feldman, D.E.; Chen, C.; Punj, V.; Machida, K. The TBC1D15 oncoprotein controls stem cell self-renewal through destabilization of the Numb-p53 complex. PLoS ONE 2013, 8, e57312.
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616.
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713.
- Cope, K.; Risby, T.; Diehl, A.M. Increased gastrointestinal ethanol production in obese mice: Implications for fatty liver disease pathogenesis. Gastroenterology 2000, 119, 1340–1347.
- Kwong, T.N.Y.; Wang, X.; Nakatsu, G.; Chow, T.C.; Tipoe, T.; Dai, R.Z.W.; Tsoi, K.K.K.; Wong, M.C.S.; Tse, G.; Chan, M.T.V.; et al. Association Between Bacteremia From Specific Microbes and Subsequent Diagnosis of Colorectal Cancer. Gastroenterology 2018, 155, 383–390.e8.
- Brown, J.M.; Hazen, S.L. The gut microbial endocrine organ: Bacterially derived signals driving cardiometabolic diseases. Annu. Rev. Med. 2015, 66, 343–359.
- Tanoue, T.; Morita, S.; Plichta, D.R.; Skelly, A.N.; Suda, W.; Sugiura, Y.; Narushima, S.; Vlamakis, H.; Motoo, I.; Sugita, K.; et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 2019, 565, 600–605.
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet 2015, 47, 505–511.
- Castaneda, F.E.; Walia, B.; Vijay-Kumar, M.; Patel, N.R.; Roser, S.; Kolachala, V.L.; Rojas, M.; Wang, L.; Oprea, G.; Garg, P.; et al. Targeted deletion of metalloproteinase 9 attenuates experimental colitis in mice: Central role of epithelial-derived MMP. Gastroenterology 2005, 129, 1991–2008.
- Reynolds, J.M.; Pappu, B.P.; Peng, J.; Martinez, G.J.; Zhang, Y.; Chung, Y.; Ma, L.; Yang, X.O.; Nurieva, R.I.; Tian, Q.; et al. Toll-like receptor 2 signaling in CD4(+) T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity 2010, 32, 692–702.
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588.
- Frosali, S.; Pagliari, D.; Gambassi, G.; Landolfi, R.; Pandolfi, F.; Cianci, R. How the Intricate Interaction among Toll-Like Receptors, Microbiota, and Intestinal Immunity Can Influence Gastrointestinal Pathology. J. Immunol. Res. 2015, 2015, 489821.
- Spiljar, M.; Merkler, D.; Trajkovski, M. The Immune System Bridges the Gut Microbiota with Systemic Energy Homeostasis: Focus on TLRs, Mucosal Barrier, and SCFAs. Front. Immunol. 2017, 8, 1353.
- Malik, S.; Sadhu, S.; Elesela, S.; Pandey, R.P.; Chawla, A.S.; Sharma, D.; Panda, L.; Rathore, D.; Ghosh, B.; Ahuja, V.; et al. Transcription factor Foxo1 is essential for IL-9 induction in T helper cells. Nat. Commun. 2017, 8, 815.
- Negi, V.; Paul, D.; Das, S.; Bajpai, P.; Singh, S.; Mukhopadhyay, A.; Agrawal, A.; Ghosh, B. Altered expression and editing of miRNA-100 regulates iTreg differentiation. Nucleic Acids Res. 2015, 43, 8057–8065.
- Atarashi, K.; Tanoue, T.; Ando, M.; Kamada, N.; Nagano, Y.; Narushima, S.; Suda, W.; Imaoka, A.; Setoyama, H.; Nagamori, T.; et al. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 2015, 163, 367–380.
- Goto, Y.; Panea, C.; Nakato, G.; Cebula, A.; Lee, C.; Diez, M.G.; Laufer, T.M.; Ignatowicz, L.; Ivanov, I.I. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity 2014, 40, 594–607.
- Wu, W.; Liu, H.P.; Chen, F.; Liu, H.; Cao, A.T.; Yao, S.; Sun, M.; Evans-Marin, H.L.; Zhao, Y.; Zhao, Q.; et al. Commensal A4 bacteria inhibit intestinal Th2-cell responses through induction of dendritic cell TGF-beta production. Eur. J. Immunol. 2016, 46, 1162–1167.
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341.
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236.
- Hrncir, T.; Stepankova, R.; Kozakova, H.; Hudcovic, T.; Tlaskalova-Hogenova, H. Gut microbiota and lipopolysaccharide content of the diet influence development of regulatory T cells: Studies in germ-free mice. BMC Immunol. 2008, 9, 65.
- Telesford, K.M.; Yan, W.; Ochoa-Reparaz, J.; Pant, A.; Kircher, C.; Christy, M.A.; Begum-Haque, S.; Kasper, D.L.; Kasper, L.H. A commensal symbiotic factor derived from Bacteroides fragilis promotes human CD39(+)Foxp3(+) T cells and Treg function. Gut Microbes 2015, 6, 234–242.
- Neff, C.P.; Rhodes, M.E.; Arnolds, K.L.; Collins, C.B.; Donnelly, J.; Nusbacher, N.; Jedlicka, P.; Schneider, J.M.; McCarter, M.D.; Shaffer, M.; et al. Diverse Intestinal Bacteria Contain Putative Zwitterionic Capsular Polysaccharides with Anti-inflammatory Properties. Cell Host Microbe 2016, 20, 535–547.
- Stingele, F.; Corthesy, B.; Kusy, N.; Porcelli, S.A.; Kasper, D.L.; Tzianabos, A.O. Zwitterionic polysaccharides stimulate T cells with no preferential V beta usage and promote anergy, resulting in protection against experimental abscess formation. J. Immunol. 2004, 172, 1483–1490.
- Dasgupta, S.; Erturk-Hasdemir, D.; Ochoa-Reparaz, J.; Reinecker, H.C.; Kasper, D.L. Plasmacytoid dendritic cells mediate anti-inflammatory responses to a gut commensal molecule via both innate and adaptive mechanisms. Cell Host Microbe 2014, 15, 413–423.
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005, 122, 107–118.
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180.
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65.
- Tzianabos, A.O.; Pantosti, A.; Baumann, H.; Brisson, J.R.; Jennings, H.J.; Kasper, D.L. The capsular polysaccharide of Bacteroides fragilis comprises two ionically linked polysaccharides. J. Biol. Chem. 1992, 267, 18230–18235.
- Cebula, A.; Seweryn, M.; Rempala, G.A.; Pabla, S.S.; McIndoe, R.A.; Denning, T.L.; Bry, L.; Kraj, P.; Kisielow, P.; Ignatowicz, L. Thymus-derived regulatory T cells contribute to tolerance to commensal microbiota. Nature 2013, 497, 258–262.
- Sonnenburg, J.L.; Chen, C.T.; Gordon, J.I. Genomic and metabolic studies of the impact of probiotics on a model gut symbiont and host. PLoS Biol. 2006, 4, e413.
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G.; et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 675–688.e7.
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141.
- Llopis, M.; Cassard, A.M.; Wrzosek, L.; Boschat, L.; Bruneau, A.; Ferrere, G.; Puchois, V.; Martin, J.C.; Lepage, P.; Le Roy, T.; et al. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut 2016, 65, 830–839.
- Chatterjee, A.; Johnson, C.N.; Luong, P.; Hullahalli, K.; McBride, S.W.; Schubert, A.M.; Palmer, K.L.; Carlson, P.E., Jr.; Duerkop, B.A. Bacteriophage Resistance Alters Antibiotic-Mediated Intestinal Expansion of Enterococci. Infect. Immun. 2019, 87, e00085-19.
- Duan, Y.; Llorente, C.; Lang, S.; Brandl, K.; Chu, H.; Jiang, L.; White, R.C.; Clarke, T.H.; Nguyen, K.; Torralba, M.; et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 2019, 575, 505–511.
- Woodhouse, C.A.; Patel, V.C.; Singanayagam, A.; Shawcross, D.L. Review article: The gut microbiome as a therapeutic target in the pathogenesis and treatment of chronic liver disease. Aliment Pharmacol. Ther. 2018, 47, 192–202.
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64.
- Kim, H.J.; Lee, J.; Choi, J.H.; Bahinski, A.; Ingber, D.E. Co-culture of Living Microbiome with Microengineered Human Intestinal Villi in a Gut-on-a-Chip Microfluidic Device. J. Vis. Exp. 2016, 114, e54344.
- De Gregorio, V.; Telesco, M.; Corrado, B.; Rosiello, V.; Urciuolo, F.; Netti, P.A.; Imparato, G. Intestine-Liver Axis On-Chip Reveals the Intestinal Protective Role on Hepatic Damage by Emulating Ethanol First-Pass Metabolism. Front. Bioeng. Biotechnol. 2020, 8, 163.
- Kasendra, M.; Luc, R.; Yin, J.; Manatakis, D.V.; Kulkarni, G.; Lucchesi, C.; Sliz, J.; Apostolou, A.; Sunuwar, L.; Obrigewitch, J.; et al. Duodenum Intestine-Chip for preclinical drug assessment in a human relevant model. eLife 2020, 9, e50135.
- Grassart, A.; Malarde, V.; Gobaa, S.; Sartori-Rupp, A.; Kerns, J.; Karalis, K.; Marteyn, B.; Sansonetti, P.; Sauvonnet, N. Bioengineered Human Organ-on-Chip Reveals Intestinal Microenvironment and Mechanical Forces Impacting Shigella Infection. Cell Host Microbe 2019, 26, 435–444.
- Jang, K.J.; Otieno, M.A.; Ronxhi, J.; Lim, H.K.; Ewart, L.; Kodella, K.R.; Petropolis, D.B.; Kulkarni, G.; Rubins, J.E.; Conegliano, D.; et al. Reproducing human and cross-species drug toxicities using a Liver-Chip. Sci. Transl. Med. 2019, 11, eaax5516.
- Koszewicz, M.; Jaroch, J.; Brzecka, A.; Ejma, M.; Budrewicz, S.; Mikhaleva, L.M.; Muresanu, C.; Schield, P.; Somasundaram, S.G.; Kirkland, C.E.; et al. Dysbiosis is one of the risk factor for stroke and cognitive impairment and potential target for treatment. Pharmacol. Res. 2021, 164, 105277.
- Van den Broek, L.A.; Lloyd, R.M.; Beldman, G.; Verdoes, J.C.; McCleary, B.V.; Voragen, A.G. Cloning and characterization of arabinoxylan arabinofuranohydrolase-D3 (AXHd3) from Bifidobacterium adolescentis DSM20083. Appl. Microbiol. Biotechnol. 2005, 67, 641–647.
- Rios-Covian, D.; Arboleya, S.; Hernandez-Barranco, A.M.; Alvarez-Buylla, J.R.; Ruas-Madiedo, P.; Gueimonde, M.; de los Reyes-Gavilan, C.G. Interactions between Bifidobacterium and Bacteroides species in cofermentations are affected by carbon sources, including exopolysaccharides produced by bifidobacteria. Appl. Environ. Microbiol. 2013, 79, 7518–7524.
- Frankel, A.E.; Coughlin, L.A.; Kim, J.; Froehlich, T.W.; Xie, Y.; Frenkel, E.P.; Koh, A.Y. Metagenomic Shotgun Sequencing and Unbiased Metabolomic Profiling Identify Specific Human Gut Microbiota and Metabolites Associated with Immune Checkpoint Therapy Efficacy in Melanoma Patients. Neoplasia 2017, 19, 848–855.
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97.
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108.
- Miller, T.L.; Wolin, M.J. Pathways of acetate, propionate, and butyrate formation by the human fecal microbial flora. Appl. Environ. Microbiol. 1996, 62, 1589–1592.
- Bergman, E.N. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 1990, 70, 567–590.
- Macfarlane, S.; Macfarlane, G.T. Regulation of short-chain fatty acid production. Proc. Nutr. Soc. 2003, 62, 67–72.
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227.
- Stumpff, F. A look at the smelly side of physiology: Transport of short chain fatty acids. Pflug. Arch. 2018, 470, 571–598.
- Bajaj, J.S.; Khoruts, A. Microbiota changes and intestinal microbiota transplantation in liver diseases and cirrhosis. J. Hepatol. 2020, 72, 1003–1027.
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojarvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.
- Craven, L.; Rahman, A.; Nair Parvathy, S.; Beaton, M.; Silverman, J.; Qumosani, K.; Hramiak, I.; Hegele, R.; Joy, T.; Meddings, J.; et al. Allogenic Fecal Microbiota Transplantation in Patients With Nonalcoholic Fatty Liver Disease Improves Abnormal Small Intestinal Permeability: A Randomized Control Trial. Am. J. Gastroenterol. 2020, 115, 1055–1065.
- Liu, R.; Kang, J.D.; Sartor, R.B.; Sikaroodi, M.; Fagan, A.; Gavis, E.A.; Zhou, H.; Hylemon, P.B.; Herzog, J.W.; Li, X.; et al. Neuroinflammation in Murine Cirrhosis Is Dependent on the Gut Microbiome and Is Attenuated by Fecal Transplant. Hepatology 2020, 71, 611–626.
- Bajaj, J.S.; Salzman, N.; Acharya, C.; Takei, H.; Kakiyama, G.; Fagan, A.; White, M.B.; Gavis, E.A.; Holtz, M.L.; Hayward, M.; et al. Microbial functional change is linked with clinical outcomes after capsular fecal transplant in cirrhosis. JCI Insight 2019, 4, e133410.
- Bajaj, J.S.; Gavis, E.A.; Fagan, A.; Wade, J.B.; Thacker, L.R.; Fuchs, M.; Patel, S.; Davis, B.; Meador, J.; Puri, P.; et al. A Randomized Clinical Trial of Fecal Microbiota Transplant for Alcohol Use Disorder. Hepatology 2020, 73, 1688–1700.
- Arfianti, A.; Pok, S.; Barn, V.; Haigh, W.G.; Yeh, M.M.; Ioannou, G.N.; Teoh, N.C.; Farrell, G.C. Exercise retards hepatocarcinogenesis in obese mice independently of weight control. J. Hepatol. 2020, 73, 140–148.
This entry is offline, you can click here to edit this entry!