Ubiquitination (the covalent attachment of ubiquitin molecules to target proteins) is one of the main post-translational modifications of proteins. Historically, the type of polyubiquitination, which involves K48 lysine residues of the monomeric ubiquitin, was the first studied type of ubiquitination. It usually targets proteins for their subsequent proteasomal degradation. All the other types of ubiquitination, including monoubiquitination; multi-monoubiquitination; and polyubiquitination involving lysine residues K6, K11, K27, K29, K33, and K63 and N-terminal methionine, were defined as atypical ubiquitination (AU). Good evidence now exists that AUs, participating in the regulation of various cellular processes, are crucial for the development of Parkinson’s disease (PD). These AUs target various proteins involved in PD pathogenesis. The K6-, K27-, K29-, and K33-linked polyubiquitination of alpha-synuclein, the main component of Lewy bodies, and DJ-1 (another PD-associated protein) is involved in the formation of insoluble aggregates. Multifunctional protein kinase LRRK2 essential for PD is subjected to K63- and K27-linked ubiquitination. Mitophagy mediated by the ubiquitin ligase parkin is accompanied by K63-linked autoubiquitination of parkin itself and monoubiquitination and polyubiquitination of mitochondrial proteins with the formation of both classical K48-linked ubiquitin chains and atypical K6-, K11-, K27-, and K63-linked polyubiquitin chains. The ubiquitin-specific proteases USP30, USP33, USP8, and USP15, removing predominantly K6-, K11-, and K63-linked ubiquitin conjugates, antagonize parkin-mediated mitophagy.
- ubiquitin–proteasome system
- atypical ubiquitination
- deubiquitinase inhibitors
- Parkinson’s disease
1. Introduction
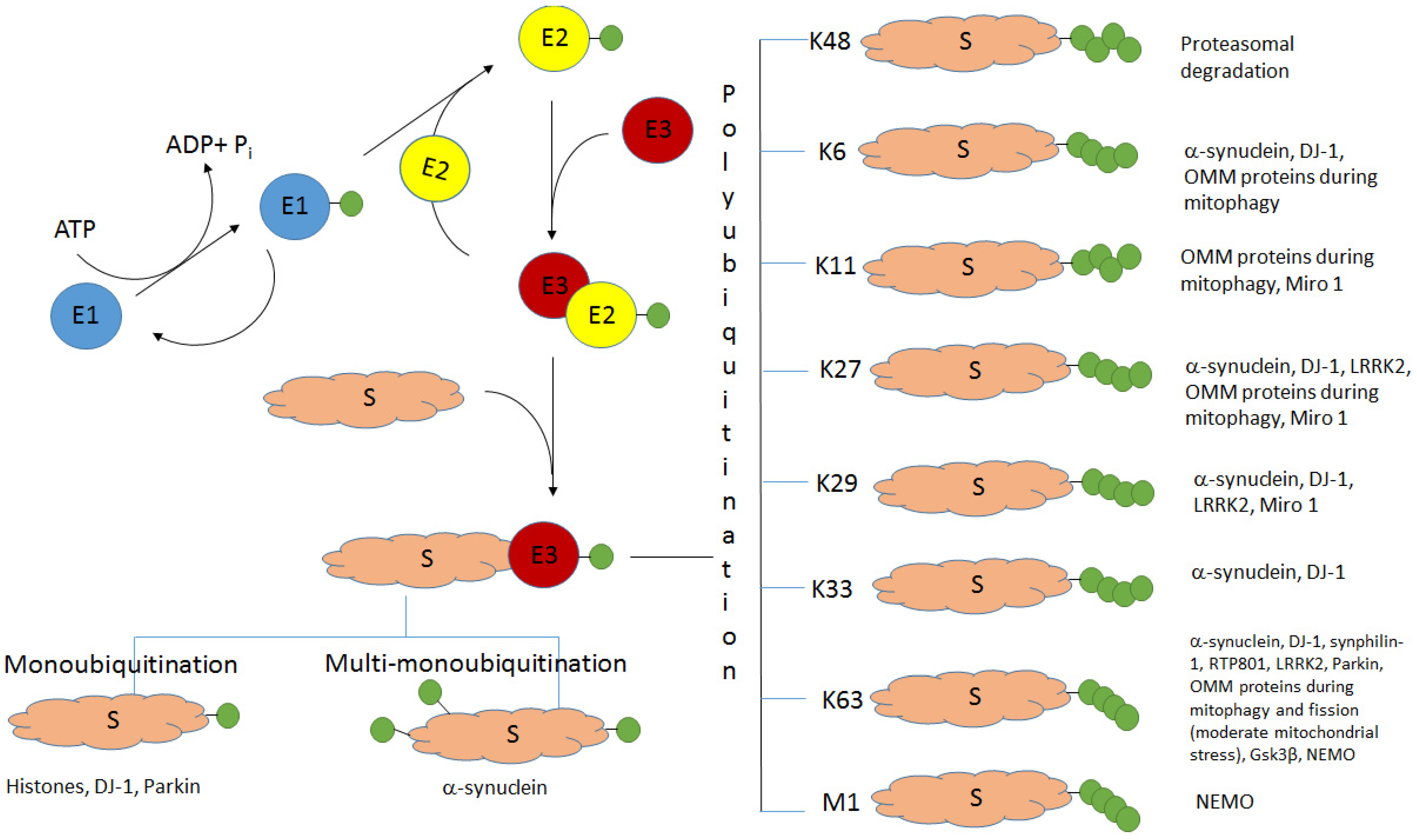

2. Alpha-Synuclein and Histones: Monoubiquitination and Multi-Monoubiquitination
3. Atypical Ubiquitination of the Components of Lewy Bodies: Alpha-Synuclein, DJ-1, and Synphilin-1
3.1. E3 Ubiquitin Ligase TRAF6: K6, K27, and K29 Ubiquitination of Alpha-Synuclein
3.2. Concerted Action of the E3 Ubiquitin Ligase Parkin with the E2 Enzyme UbcH13/Uev1a: K63-Linked Ubiquitination of Alpha-Synuclein and Synphilin-1 Promotes Lewy Body Formation
3.3. HECT E3 Ligase NEDD4: K63-Linked Ubiquitination of Alpha-Synuclein
3.4. DJ-1: Monoubiquitination and K63-Linked Polyubiquitination. DJ-1 and Alpha-Synuclein: K6-, K27-, and K29-Linked Polyubiquitination
4. LRRK2: K63- and K27-Linked Ubiquitination
5. Concerted Mechanisms of Different Types of Ubiquitination and Deubiquitination in Mitophagy (Monoubiquitination and K6-, K11-, K27-, and K63-Linked Polyubiquitination)
6. Deubiquitinases (USP8, USP15, USP30, USP33, USP35, UCH-L1) Removing Atypical Ubiquitin Conjugates from PD-Related Proteins
7. M1-Linked Ubiquitination
This entry is adapted from the peer-reviewed paper 10.3390/ijms23073705
References
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479.
- Kulathu, Y.; Komander, D. Atypical ubiquitylation—The unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat. Rev. Mol. Cell Biol. 2012, 13, 508–523.
- Sadowski, M.; Suryadinata, R.; Tan, A.R.; Roesley, S.N.; Sarcevic, B. Protein monoubiquitination and polyubiquitination generate structural diversity to control distinct biological processes. IUBMB Life 2012, 64, 136–142.
- Li, W.; Ye, Y. Polyubiquitin chains: Functions, structures, and mechanisms. Cell Mol. Life Sci. 2008, 65, 2397–2406.
- Varadan, R.; Walker, O.; Pickart, C.; Fushman, D. Structural properties of polyubiquitin chains in solution. J. Mol. Biol. 2002, 6, 637–647.
- Varadan, R.; Assfalg, M.; Haririnia, A.; Raasi, S.; Pickart, C.; Fushman, D. Solution conformation of Lys63-linked di-ubiquitin chain provides clues to functional diversity of polyubiquitin signaling. J. Biol. Chem. 2004, 279, 7055–7063.
- Hospenthal, M.K.; Freund, S.M.; Komander, D. Assembly, analysis and architecture of atypical ubiquitin chains. Nat. Struct. Mol. Biol. 2013, 20, 555–565.
- Datta, A.B.; Hura, G.L.; Wolberger, C. The structure and conformation of Lys63-linked tetraubiquitin. J. Mol. Biol. 2009, 392, 1117–1124.
- Ikeda, F.; Dikic, I. Atypical ubiquitin chains: New molecular signals. ‘Protein Modifications: Beyond the Usual Suspects’ review series. EMBO Rep. 2008, 9, 536–542.
- Walczak, H.; Iwai, K.; Dikic, I. Generation and physiological roles of linear ubiquitin chains. BMC Biol. 2012, 10, 23.
- Rittinger, K.; Ikeda, F. Linear ubiquitin chains: Enzymes, mechanisms and biology. Open. Biol. 2017, 7, 170026.
- Buneeva, O.A.; Medvedev, A.E. The role of atypical ubiquitination in cell regulation. Biochem. (Moscow) Suppl. Ser. B 2017, 11, 16–31.
- Kliza, K.; Husnjak, K. Resolving the Complexity of Ubiquitin Networks. Front. Mol. Biosci. 2020, 7, 21.
- Tenno, T.; Fujiwara, K.; Tochio, H.; Iwai, K.; Morita, E.H.; Hayashi, H.; Murata, S.; Hiroaki, H.; Sato, M.; Tanaka, K.; et al. Structural basis for distinct roles of Lys63- and Lys48-linked polyubiquitin chains. Genes Cells 2004, 9, 865–875.
- Raasi, S.; Varadan, R.; Fushman, D.; Pickart, C.M. Diverse polyubiquitin interaction properties of ubiquitin-associated domains. Nat. Struct. Mol. Biol. 2005, 12, 708–714.
- Di Fiore, P.P.; Polo, S.; Hofmann, K. When ubiquitin meets ubiquitin receptors: A signalling connection. Nat. Rev. Mol. Cell Biol. 2003, 4, 491–497.
- Samant, R.S.; Livingston, C.M.; Sontag, E.M.; Frydman, J. Distinct proteostasis circuits cooperate in nuclear and cytoplasmic protein quality control. Nature 2018, 563, 407–411.
- Aguilar, R.C.; Wendland, B. Ubiquitin: Not just for proteasomes anymore. Curr. Opin. Cell Biol. 2003, 15, 184–190.
- Tracz, M.; Bialek, W. Beyond K48 and K63: Non-canonical protein ubiquitination. Cell Mol. Biol. Lett. 2021, 26, 1.
- Huang, Q.; Zhang, X. Emerging Roles and Research Tools of Atypical Ubiquitination. Proteomics 2020, 20, e1900100.
- Nathan, J.A.; Kim, H.T.; Ting, L.; Gygi, S.P.; Goldberg, A.L. Why do cellular proteins linked to K63-polyubiquitin chains not associate with proteasomes? EMBO J. 2013, 32, 552–565.
- Martinez-Fonts, K.; Davis, C.; Tomita, T.; Elsasser, S.; Nager, A.R.; Shi, Y.; Finley, D.; Matouschek, A. The proteasome 19S cap and its ubiquitin receptors provide a versatile recognition platform for substrates. Nat. Commun. 2020, 11, 477.
- Le Guerroué, F.; Youle, R.J. Ubiquitin signaling in neurodegenerative diseases: An autophagy and proteasome perspective. Cell Death Differ. 2021, 28, 439–454.
- Fouka, M.; Mavroeidi, P.; Tsaka, G.; Xilouri, M. In Search of Effective Treatments Targeting α-Synuclein Toxicity in Synucleinopathies: Pros and Cons. Front. Cell Dev. Biol. 2020, 8, 559791.
- Burré, J.; Sharma, M.; Südhof, T.C. Cell Biology and Pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091.
- Rott, R.; Szargel, R.; Haskin, J.; Bandopadhyay, R.; Lees, A.J.; Shani, V.; Engelender, S. α-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 18666–18671.
- Livneh, I.; Kravtsova-Ivantsiv, Y.; Braten, O.; Kwon, Y.T.; Ciechanover, A. Monoubiquitination joins polyubiquitination as an esteemed proteasomal targeting signal. Bioessays 2017, 39, 1700027.
- Braten, O.; Livneh, I.; Ziv, T.; Admon, A.; Kehat, I.; Caspi, L.H.; Gonen, H.; Bercovich, B.; Godzik, A.; Jahandideh, S.; et al. Numerous proteins with unique characteristics are degraded by the 26S proteasome following monoubiquitination. Proc. Natl. Acad. Sci. USA 2016, 113, E4639–E4647.
- Liani, E.; Eyal, A.; Avraham, E.; Shemer, R.; Szargel, R.; Berg, D.; Bornemann, A.; Riess, O.; Ross, C.A.; Rott, R.; et al. Ubiquitylation of synphilin-1 and alpha-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 5500–5505.
- Rott, R.; Szargel, R.; Haskin, J.; Shani, V.; Shainskaya, A.; Manov, I.; Liani, E.; Avraham, E.; Engelender, S. Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 2008, 283, 3316–3328.
- Lee, J.T.; Wheeler, T.C.; Li, L.; Chin, L.S. Ubiquitination of alpha-synuclein by Siah-1 promotes alpha-synuclein aggregation and apoptotic cell death. Hum. Mol. Genet. 2008, 17, 906–917.
- Moon, S.P.; Balana, A.T.; Galesic, A.; Rakshit, A.; Pratt, M.R. Ubiquitination Can Change the Structure of the α-Synuclein Amyloid Fiber in a Site Selective Fashion. J. Org. Chem. 2020, 85, 1548–1555.
- Szargel, R.; Rott, R.; Eyal, A.; Haskin, J.; Shani, V.; Balan, L.; Wolosker, H.; Engelender, S. Synphilin-1A inhibits seven in absentia homolog (SIAH) and modulates alpha-synuclein monoubiquitylation and inclusion formation. J. Biol. Chem. 2009, 284, 11706–11716.
- Abeywardana, T.; Lin, Y.H.; Rott, R.; Engelender, S.; Pratt, M.R. Site-specific differences in proteasome-dependent degradation of monoubiquitinated α-synuclein. Chem. Biol. 2013, 20, 1207–1213.
- Wong, Y.C.; Krainc, D. α-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13.
- Schmidt, M.F.; Gan, Z.Y.; Komander, D.; Dewson, G. Ubiquitin signalling in neurodegeneration: Mechanisms and therapeutic opportunities. Cell Death Differ. 2021, 28, 570–590.
- Hogan, A.K.; Foltz, D.R. Reduce, Retain, Recycle: Mechanisms for Promoting Histone Protein Degradation versus Stability and Retention. Mol. Cell Biol. 2021, 41, e0000721.
- Busch, H.; Goldknopf, I.L. Ubiquitin-protein conjugates. Mol. Cell Biochem. 1981, 40, 173–187.
- Spencer, V.A.; Davie, J.R. Role of covalent modifications of histones in regulating gene expression. Gene 1999, 240, 1–12.
- Zhou, W.; Zhu, P.; Wang, J.; Pascual, G.; Ohgi, K.A.; Lozach, J.; Glass, C.K.; Rosenfeld, M.G. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol. Cell 2008, 29, 69–80.
- Cao, J.; Yan, Q. Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front. Oncol. 2012, 2, 26.
- Srivastava, A.; McGrath, B.; Bielas, S.L. Histone H2A Monoubiquitination in Neurodevelopmental Disorders. Trends Genet. 2017, 33, 566–578.
- Stewart, M.D.; Zelin, E.; Dhall, A.; Walsh, T.; Upadhyay, E.; Corn, J.E.; Chatterjee, C.; King, M.C.; Klevit, R.E. BARD1 is necessary for ubiquitylation of nucleosomal histone H2A and for transcriptional regulation of estrogen metabolism genes. Proc. Natl. Acad. Sci. USA 2018, 115, 1316–1321.
- Rape, M. Ubiquitylation at the crossroads of development and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 59–70.
- Lim, K.H.; Song, M.H.; Baek, K.H. Decision for cell fate: Deubiquitinating enzymes in cell cycle checkpoint. Cell Mol. Life Sci. 2016, 73, 1439–1455.
- Ben Yehuda, A.; Risheq, M.; Novoplansky, O.; Bersuker, K.; Kopito, R.R.; Goldberg, M.; Brandeis, M. Ubiquitin Accumulation on Disease Associated Protein Aggregates Is Correlated with Nuclear Ubiquitin Depletion, Histone De-Ubiquitination and Impaired DNA Damage Response. PLoS ONE 2017, 12, e0169054.
- Aquila, L.; Atanassov, B.S. Regulation of Histone Ubiquitination in Response to DNA Double Strand Breaks. Cells 2020, 9, 1699.
- Bonnet, J.; Devys, D.; Tora, L. Histone H2B ubiquitination: Signaling not scrapping. Drug Discov. Today Technol. 2014, 12, e19–e27.
- Wu, Y.; Chen, P.; Jing, Y.; Wang, C.; Men, Y.L.; Zhan, W.; Wang, Q.; Gan, Z.; Huang, J.; Xie, K.; et al. Microarray Analysis Reveals Potential Biological Functions of Histone H2B Monoubiquitination. PLoS ONE 2015, 10, e0133444.
- Marsh, D.J.; Ma, Y.; Dickson, K.A. Histone Monoubiquitination in Chromatin Remodelling: Focus on the Histone H2B Interactome and Cancer. Cancers 2020, 12, 3462.
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282.
- Maynard, S.; Fang, E.F.; Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. DNA Damage, DNA Repair, Aging, and Neurodegeneration. Cold Spring Harb. Perspect. Med. 2015, 5, a025130.
- Vissers, J.H.; Nicassio, F.; van Lohuizen, M.; Di Fiore, P.P.; Citterio, E. The many faces of ubiquitinated histone H2A: Insights from the DUBs. Cell Div. 2008, 3, 8.
- Scheuermann, J.C.; Gutiérrez, L.; Müller, J. Histone H2A monoubiquitination and Polycomb repression: The missing pieces of the puzzle. Fly 2012, 6, 162–168.
- Peña-Altamira, L.E.; Polazzi, E.; Monti, B. Histone post-translational modifications in Huntington’s and Parkinson’s diseases. Curr. Pharm. Des. 2013, 19, 5085–5092.
- Zucchelli, S.; Codrich, M.; Marcuzzi, F.; Pinto, M.; Vilotti, S.; Biagioli, M.; Ferrer, I.; Gustincich, S. TRAF6 promotes atypical ubiquitination of mutant DJ-1 and alpha-synuclein and is localized to Lewy bodies in sporadic Parkinson’s disease brains. Hum. Mol. Genet. 2010, 19, 3759–3770.
- Doss-Pepe, E.W.; Chen, L.; Madura, K. Alpha-synuclein and parkin contribute to the assembly of ubiquitin lysine 63-linked multiubiquitin chains. J. Biol. Chem. 2005, 280, 16619–16624.
- Lim, K.L.; Chew, K.C.; Tan, J.M.; Wang, C.; Chung, K.K.; Zhang, Y.; Tanaka, Y.; Smith, W.; Engelender, S.; Ross, C.A.; et al. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: Implications for Lewy body formation. J. Neurosci. 2005, 25, 2002–2009.
- Lim, K.L.; Dawson, V.L.; Dawson, T.M. Parkin-mediated lysine 63-linked polyubiquitination: A link to protein inclusions formation in Parkinson’s and other conformational diseases? Neurobiol. Aging 2006, 27, 524–529.
- Tofaris, G.K.; Kim, H.T.; Hourez, R.; Jung, J.W.; Kim, K.P.; Goldberg, A.L. Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 17004–17009.
- Boassa, D.; Berlanga, M.L.; Yang, M.A.; Terada, M.; Hu, J.; Bushong, E.A.; Hwang, M.; Masliah, E.; George, J.M.; Ellisman, M.H. Mapping the subcellular distribution of α-synuclein in neurons using genetically encoded probes for correlated light and electron microscopy: Implications for Parkinson’s disease pathogenesis. J. Neurosci. 2013, 33, 2605–2615.
- Sugeno, N.; Hasegawa, T.; Tanaka, N.; Fukuda, M.; Wakabayashi, K.; Oshima, R.; Konno, M.; Miura, E.; Kikuchi, A.; Baba, T.; et al. Lys-63-linked ubiquitination by E3 ubiquitin ligase Nedd4-1 facilitates endosomal sequestration of internalized α-synuclein. J. Biol. Chem. 2014, 289, 18137–18151.
- Buneeva, O.A.; Medvedev, A.E. DJ-1 Protein and Its Role in the Development of Parkinson’s Disease: Studies on Experimental Models. Biochemistry 2021, 86, 627–640.
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188.
- Mencke, P.; Boussaad, I.; Romano, C.D.; Kitami, T.; Linster, C.L.; Krüger, R. The Role of DJ-1 in Cellular Metabolism and Pathophysiological Implications for Parkinson’s Disease. Cells 2021, 10, 347.
- Ramsey, C.P.; Giasson, B.I. L10p and P158DEL DJ-1 mutations cause protein instability, aggregation, and dimerization impairments. J. Neurosci. Res. 2010, 88, 3111–3124.
- Moore, D.J.; Zhang, L.; Dawson, T.M.; Dawson, V.L. A missense mutation (L166P) in DJ-1, linked to familial Parkinson’s disease, confers reduced protein stability and impairs homo-oligomerization. J. Neurochem. 2003, 87, 1558–1567.
- Scumaci, D.; Olivo, E.; Fiumara, C.V.; La Chimia, M.; De Angelis, M.T.; Mauro, S.; Costa, G.; Ambrosio, F.A.; Alcaro, S.; Agosti, V.; et al. DJ-1 proteoforms in breast cancer cells: The escape of metabolic epigenetic misregulation. Cells 2020, 9, 1968.
- Junn, E.; Taniguchi, H.; Jeong, B.S.; Zhao, X.; Ichijo, H.; Mouradian, M.M. Interaction of DJ-1 with Daxx inhibits apoptosis signal-regulating kinase 1 activity and cell death. Proc. Natl. Acad. Sci. USA 2005, 102, 9691–9696.
- Takahashi, K.; Taira, T.; Niki, T.; Seino, C.; Iguchi-Ariga, S.M.; Ariga, H. DJ-1 positively regulates the androgen receptor by impairing the binding of PIASx alpha to the receptor. J. Biol. Chem. 2001, 276, 37556–37563.
- Niki, T.; Takahashi-Niki, K.; Taira, T.; Iguchi-Ariga, S.M.; Ariga, H. DJBP: A novel DJ-1-binding protein, negatively regulates the androgen receptor by recruiting histone deacetylase complex, and DJ-1 antagonizes this inhibition by abrogation of this complex. Mol. Cancer Res. 2003, 1, 247–261.
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096.
- Shinbo, Y.; Taira, T.; Niki, T.; Iguchi-Ariga, S.M.; Ariga, H. DJ-1 restores p53 transcription activity inhibited by Topors/p53BP3. Int. J. Oncol. 2005, 26, 641–648.
- Kato, I.; Maita, H.; Takahashi-Niki, K.; Saito, Y.; Noguchi, N.; Iguchi-Ariga, S.M.; Ariga, H. Oxidized DJ-1 inhibits p53 by sequestering p53 from promoters in a DNA-binding affinity-dependent manner. Mol. Cell. Biol. 2013, 33, 340–359.
- Zhong, N.; Kim, C.Y.; Rizzu, P.; Geula, C.; Porter, D.R.; Pothos, E.N.; Squitieri, F.; Heutink, P.; Xu, J. DJ-1 transcriptionally up-regulates the human tyrosine hydroxylase by inhibiting the sumoylation of pyrimidine tractbinding protein-associated splicing factor. J. Biol. Chem. 2006, 281, 20940–20948.
- Ishikawa, S.; Taira, T.; Takahashi-Niki, K.; Niki, T.; Ariga, H.; Iguchi-Ariga, S.M. Human DJ-1-specific transcriptional activation of tyrosine hydroxylase gene. J. Biol. Chem. 2010, 285, 39718–39731.
- Shinbo, Y.; Niki, T.; Taira, T.; Ooe, H.; Takahashi-Niki, K.; Maita, C.; Seino, C.; Iguchi-Ariga, S.M.; Ariga, H. Proper SUMO-1 conjugation is essential to DJ-1 to exert its full activities. Cell Death Differ. 2006, 13, 96–108.
- Ariga, H.; Takahashi-Niki, K.; Kato, I.; Maita, H.; Niki, T.; Iguchi-Ariga, S.M. Neuroprotective function of DJ-1 in Parkinson’s disease. Oxid. Med. Cell Longev. 2013, 2013, 683920.
- Xiong, H.; Wang, D.; Chen, L.; Choo, Y.S.; Ma, H.; Tang, C.; Xia, K.; Jiang, W.; Ronai, Z.; Zhuang, X.; et al. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J. Clin. Investig. 2009, 119, 650–660.
- Olzmann, J.A.; Bordelon, J.R.; Muly, E.C.; Rees, H.D.; Levey, A.I.; Li, L.; Chin, L.S. Selective enrichment of DJ-1 protein in primate striatal neuronal processes: Implications for Parkinson’s disease. J. Comp. Neurol. 2007, 500, 585–599.
- Parsanejad, M.; Zhang, Y.; Qu, D.; Irrcher, I.; Rousseaux, M.W.; Aleyasin, H.; Kamkar, F.; Callaghan, S.; Slack, R.S.; Mak, T.W.; et al. Regulation of the VHL/HIF-1 pathway by DJ-1. J. Neurosci. 2014, 34, 8043–8050.
- Lisztwan, J.; Imbert, G.; Wirbelauer, C.; Gstaiger, M.; Krek, W. The von Hippel-Lindau tumor suppressor protein is a component of an E3 ubiquitin-protein ligase activity. Genes Dev. 1999, 13, 1822–1833.
- Iwai, K.; Yamanaka, K.; Kamura, T.; Minato, N.; Conaway, R.C.; Conaway, J.W.; Klausner, R.D.; Pause, A. Identification of the von Hippel-lindau tumor-suppressor protein as part of an active E3 ubiquitin ligase complex. Proc. Natl. Acad. Sci. USA 1999, 96, 12436–12441.
- Hatcher, J.M.; Choi, H.G.; Alessi, D.R.; Gray, N.S. Small-Molecule Inhibitors of LRRK2. Adv. Neurobiol. 2017, 14, 241–264.
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107.
- Jeong, G.R.; Lee, B.D. Pathological Functions of LRRK2 in Parkinson’s Disease. Cells 2020, 9, 2565.
- Ko, H.S.; Bailey, R.; Smith, W.W.; Liu, Z.; Shin, J.H.; Lee, Y.I.; Zhang, Y.J.; Jiang, H.; Ross, C.A.; Moore, D.J.; et al. CHIP regulates leucine-rich repeat kinase-2 ubiquitination, degradation, and toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 2897–2902.
- Liu, Y.; Sun, Y.; Han, S.; Guo, Y.; Tian, Q.; Ma, Q.; Zhang, S. CHIP promotes the activation of NF-κB signaling through enhancing the K63-linked ubiquitination of TAK1. Cell Death Discov. 2021, 7, 246.
- Buneeva, O.A.; Medvedeva, M.V.; Kopylov, A.T.; Medvedev, A.E. Ubiquitin subproteome of brain mitochondria and its changes induced by experimental Parkinsonism and action of neuroprotectors. Biochemistry 2019, 84, 1359–1374.
- Nucifora, F.C., Jr.; Nucifora, L.G.; Ng, C.H.; Arbez, N.; Guo, Y.; Roby, E.; Shani, V.; Engelender, S.; Wei, D.; Wang, X.F.; et al. Ubiqutination via K27 and K29 chains signals aggregation and neuronal protection of LRRK2 by WSB1. Nat. Commun. 2016, 7, 11792.
- Thomas, J.M.; Wang, X.; Guo, G.; Li, T.; Dai, B.; Nucifora, L.G.; Nucifora, F.C., Jr.; Liu, Z.; Xue, F.; Liu, C.; et al. GTP-binding inhibitors increase LRRK2-linked ubiquitination and Lewy body-like inclusions. J. Cell Physiol. 2020, 235, 7309–7320.
- Jenner, P.; Olanow, C.W. Understanding cell death in Parkinson’s disease. Ann. Neurol. 1998, 44 (Suppl. 1), S72–S84.
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306.
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909.
- Beal, M.F. Mitochondria, oxidative damage, and inflammation in Parkinson’s disease. Ann. N. Y. Acad. Sci. 2003, 991, 120–131.
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Dysfunction in Parkinson’s Disease—Cause or Consequence? Biology 2019, 8, 38.
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. 1), 216–231.
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells 2019, 8, 712.
- Sedlackova, L.; Korolchuk, V.I. Mitochondrial quality control as a key determinant of cell survival. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 575–587.
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 2012, 22, 320–333.
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298.
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating serine 65. Open. Biol. 2012, 2, 120080.
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153.
- Wauer, T.; Swatek, K.N.; Wagstaff, J.L.; Gladkova, C.; Pruneda, J.N.; Michel, M.A.; Gersch, M.; Johnson, C.M.; Freund, S.M.; Komander, D. Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J. 2015, 34, 307–325.
- Durcan, T.M.; Fon, E.A. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999.
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 2014, 56, 360–375.
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20.
- Cunningham, C.N.; Baughman, J.M.; Phu, L.; Tea, J.S.; Yu, C.; Coons, M.; Kirkpatrick, D.S.; Bingol, B.; Corn, J.E. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat. Cell Biol. 2015, 17, 160–169.
- Durcan, T.M.; Tang, M.Y.; Pérusse, J.R.; Dashti, E.A.; Aguileta, M.A.; McLelland, G.L.; Gros, P.; Shaler, T.A.; Faubert, D.; Coulombe, B.; et al. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J. 2014, 33, 2473–2491.
- Durcan, T.M.; Fon, E.A. USP8 and PARK2/parkin-mediated mitophagy. Autophagy 2015, 11, 428–429.
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131.
- Amer-Sarsour, F.; Kordonsky, A.; Berdichevsky, Y.; Prag, G.; Ashkenazi, A. Deubiquitylating enzymes in neuronal health and disease. Cell Death Dis. 2021, 12, 120.
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422.
- Lim, K.H.; Joo, J.Y.; Baek, K.H. The potential roles of deubiquitinating enzymes in brain diseases. Aging Res. Rev. 2020, 61, 101088.
- Wang, Y.; Serricchio, M.; Jauregui, M.; Shanbhag, R.; Stoltz, T.; Di Paolo, C.T.; Kim, P.K.; McQuibban, G.A. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 2015, 11, 595–606.
- Clague, M.J.; Urbé, S.; Komander, D. Breaking the chains: Deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol. 2019, 20, 338–352.
- Grumati, P.; Dikic, I. Ubiquitin signaling and autophagy. J. Biol. Chem. 2018, 293, 5404–5413.
- Alexopoulou, Z.; Lang, J.; Perrett, R.M.; Elschami, M.; Hurry, M.E.; Kim, H.T.; Mazaraki, D.; Szabo, A.; Kessler, B.M.; Goldberg, A.L.; et al. Deubiquitinase Usp8 regulates α-synuclein clearance and modifies its toxicity in Lewy body disease. Proc. Natl. Acad. Sci. USA 2016, 113, E4688–E4697.
- Niu, K.; Fang, H.; Chen, Z.; Zhu, Y.; Tan, Q.; Wei, D.; Li, Y.; Balajee, A.S.; Zhao, Y. USP33 deubiquitinates PRKN/parkin and antagonizes its role in mitophagy. Autophagy 2020, 16, 724–734.
- Cornelissen, T.; Haddad, D.; Wauters, F.; Van Humbeeck, C.; Mandemakers, W.; Koentjoro, B.; Sue, C.; Gevaert, K.; De Strooper, B.; Verstreken, P.; et al. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet. 2014, 23, 5227–5242.
- Rusilowicz-Jones, E.V.; Jardine, J.; Kallinos, A.; Pinto-Fernandez, A.; Guenther, F.; Giurrandino, M.; Barone, F.G.; McCarron, K.; Burke, C.J.; Murad, A.; et al. USP30 sets a trigger threshold for PINK1-PARKIN amplification of mitochondrial ubiquitylation. Life Sci. Alliance 2020, 3, e202000768.
- Ordureau, A.; Paulo, J.A.; Zhang, J.; An, H.; Swatek, K.N.; Cannon, J.R.; Wan, Q.; Komander, D.; Harper, J.W. Global Landscape and Dynamics of Parkin and USP30-Dependent Ubiquitylomes in iNeurons during Mitophagic Signaling. Mol. Cell 2020, 77, 1124–1142.e10.
- Leroy, E.; Boyer, R.; Auburger, G.; Leube, B.; Ulm, G.; Mezey, E.; Harta, G.; Brownstein, M.J.; Jonnalagada, S.; Chernova, T.; et al. The ubiquitin pathway in Parkinson’s disease. Nature 1998, 395, 451–452.
- Lee, Y.C.; Hsu, S.D. Familial Mutations and Post-translational Modifications of UCH-L1 in Parkinson’s Disease and Neurodegenerative Disorders. Curr. Protein Pept. Sci. 2017, 18, 733–745.
- Lowe, J.; McDermott, H.; Landon, M.; Mayer, R.J.; Wilkinson, K.D. Ubiquitin carboxyl-terminal hydrolase (PGP 9.5) is selectively present in ubiquitinated inclusion bodies characteristic of human neurodegenerative diseases. J. Pathol. 1990, 161, 153–160.
- Tokunaga, F. Linear ubiquitination-mediated NF-κB regulation and its related disorders. J. Biochem. 2013, 154, 313–323.
- Emmerich, C.H.; Ordureau, A.; Strickson, S.; Arthur, J.S.; Pedrioli, P.G.; Komander, D.; Cohen, P. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc. Natl. Acad. Sci. USA 2013, 110, 15247–15252.
- Hrdinka, M.; Gyrd-Hansen, M. The Met1-Linked Ubiquitin Machinery: Emerging Themes of (De)regulation. Mol. Cell 2017, 68, 265–280.
- Dittmar, G.; Winklhofer, K.F. Linear Ubiquitin Chains: Cellular Functions and Strategies for Detection and Quantification. Front. Chem. 2020, 7, 915.
- Wang, Y.; Shan, B.; Liang, Y.; Wei, H.; Yuan, J. Parkin regulates NF-κB by mediating site-specific ubiquitination of RIPK1. Cell Death Dis. 2018, 9, 732.
- Degterev, A.; Ofengeim, D.; Yuan, J. Targeting RIPK1 for the treatment of human diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 9714–9722.