Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Nanoscience & Nanotechnology
Perylene diimide (PDI) has attracted attention owing to its chemical robustness, thermal and photo-stability, and outstanding optical and electronic properties.
- perylene diimide
- self-assembly
- nanostructures
- peptides
- DNA
- Bioconjugates
1. Introduction
Mother Nature has always been a unique source of inspiration for the design of nanostructures and materials for application in various fields spanning from biomedicine to electronics. Living organisms are hierarchically built on the organized self-assembly of biomacromolecules, such as lipids, proteins, deoxyribonucleic acids (DNA), ribonucleic acids (RNA), and polysaccharides, controlled by a delicate balance of non-covalent interactions [1,2]. For example, the cellular plasma membrane results from the spontaneous organization of a diversity of lipids into a fluid and complex bilayer, while the exceptional mechanical properties of spider silk arise from the coordinated self-recognition of proteins. Over the last decades, biological macromolecules have been continuously harnessed as building blocks for the construction of (nano)structures with atomic-scale precision. Owing to their biocompatibility, functionality, and ability to undergo self-assembly, the conjugation of biological macromolecules to organic self-assembling molecules with interesting (photo)chemical properties has supported the construction of hybrid supramolecular architectures [3,4]. Of such small organic molecules, the family of perylene diimide (PDI; perylene-3,4:9,10-tetracarboxylic acid diimide) dyes has attracted tremendous attention. Particularly, PDI-based chromophores are known for their electron mobility, high fluorescence quantum yields, photo- and thermal stability, and semi-conductivity [5,6]. These properties benefit them in applications such as pigments, dye lasers, sensors, bioprobes, and photovoltaics [5,7,8]. Interestingly, the physicochemical, optical, and structural properties of PDI derivatives can be tuned by the modification of their substituents. Particularly, conjugating PDIs to biological macromolecules can allow the conception of supramolecular structures with unique architectures and properties. Albeit these chimeric PDI-biomolecules have been exploited for two decades, their self-assembly as well as the resulting morphologies remain difficult to predict from the monomeric building blocks, still precluding their full potential.
2. Synthesis and Properties of PDI Derivatives
Owing to their stability and unique optical properties, organic π-conjugated systems constitute an important class of molecules for different applications in various fields, from biomedicine to (nano)material sciences. In particular, PDI and its derivatives are π-conjugated systems that have been widely exploited as building blocks for the preparation of stable and functional nanostructures with tailored optical and physicochemical properties [5,10,11]. To this end, several groups have developed innovative and reliable methods for the efficient synthesis of PDI derivatives and their subsequent self-assembly into tailored structures. In this first section, we will offer a brief overview of the main synthetic approaches commonly used to prepare PDI and its derivatives and we will present the optical, physicochemical, and self-assembly properties of PDI-based π-conjugated systems.
2.1. Physicochemical, Optical, and Self-Assembling Properties of PDIs
PDI derivatives have been recognized as an ideal π-conjugated system for chemical, colorimetric and fluorescent sensors [12,13], organic semiconductors and optoelectronic devices [14,15,16], phototheranostics, [17,18] as well as bioimaging and gene/drug delivering agents [19]. Structurally, the PDI molecule is composed of one rigid, planar, and stable perylene core with two imide groups at both ends of the polycyclic aromatic scaffold. The perylene core has 12 characteristic positions, known as the bay (1, 6, 7 and 12 positions), ortho (2, 5, 8 and 11 positions), and peri (3, 4, 9 and 10 positions). The attractiveness of PDIs as chromophores is associated with their chemical robustness, excellent thermal and photo-stabilities as well as their unique optical and electronic properties [9,10]. Soluble monomeric PDIs without substituents on the ortho and bay positions of the perylene core show prototypical UV-Vis absorption spectra characterized with three vibronic peaks and a typical mirror image emission spectrum with high fluorescence quantum yields in most organic solvents [5,20]. Interestingly, substitutions at the two imide positions have a negligible effect on the optical properties of PDIs, while they significantly modulate the self-assembling processes as well as the architecture of the resulting supramolecular structures [11,21]. In contrast, substituents at the bay positions have a considerable effect on the morphology as well as the optical, chiroptical [22], and electronic properties of PDI-based assemblies, such as exciton diffusion, charge transfer, and high fluorescence quantum yield with red-shifted emission bands, which are associated with applications in organic solar cells [23], organic semiconductor devices [24], organic light-emitting diodes [25], vapor sensors [26,27,28], and theranostic agents [29,30]. These bay-substitutions can lead to a distortion of the planar PDI that affects solubility and weakens the π–π staking interactions [11]. Consequently, the twisting of the PDI cores can be precisely tuned through bay-substitutions, leading to some control over the self-assembly and the desired morphology of the nano- and mesoscopic structures [30] and liquid-crystalline materials [31,32].
Generally, PDIs form weakly emissive H-aggregates in aqueous solutions via non-covalent π–π stacking interactions, leading to the linear orientation of aromatic systems, i.e., one top of each other (Figure 1). In contrast, the modulation of the planar structure by introducing bulky substituents into the bay positions of the perylene core can generate strongly emissive core-twisted self-assembled supramolecular structures [5], in which neighboring chromophores are oriented in a head-to-tail fashion, known as J-aggregates (Figure 1) [33,34].
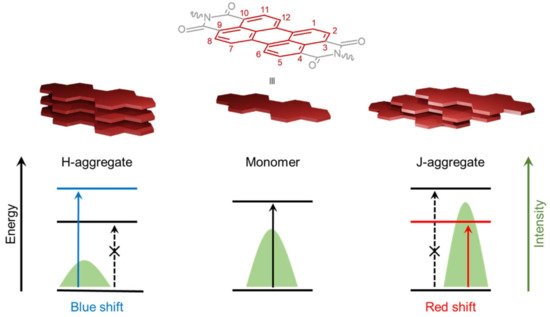
Figure 1. Molecular structure of PDI with the 12 positions of the perylene core. Arrangements of H- (left) and J- (right) aggregates resulting from the self-assembly of PDI monomers (center) with the corresponding effects on fluorescence emission spectra and electronic π–π * transitions. Full arrows indicate the permitted transitions and dashed arrows the forbidden ones.
2.2. Synthetic Strategies to Access Symmetrical PDIs
Perylene-3,4,9,10-tetracarboxylic dianhydride (PTCDA) is considered the parent compound of PDIs and is easily accessible through several well-defined multistep processes, including the one represented in Scheme 1 [20,21,35]. In this example, the first step includes the synthesis of naphthalene-1,8-dicarboxanhydride via the oxidation of acenaphthene, followed by ammonia treatment to convert the anhydride to an imide functionality. Imide dimerization facilitated by molten potassium hydroxide provides perylene-3,4,9,10-tetracarboxylic diimide (PTCDI) that can be hydrolyzed using concentrated sulfuric acid at high temperature to obtain PTCDA. Imidization of PTCDA with primary amines or anilines, in imidazole and in the presence of anhydrous zinc acetate as catalyst, leads to the formation of appropriate symmetrically N,N’-substituted PDIs in high yields [20,21,35,36].
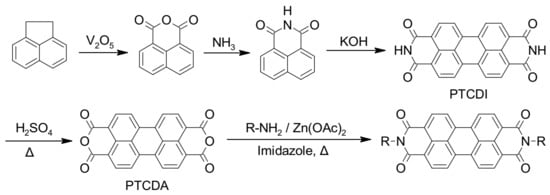
Scheme 1. Common synthetic pathway for the preparation of PTCDA and symmetrically N,N’-substituted PDIs.
2.3. Synthetic Strategies to Access Asymmetrical PDIs
Asymmetrical PDIs can be obtained by the partial hydrolysis of PDIs to perylene monoimide monoanhydride followed by condensation of this mixed imide–anhydride compound with primary amines to access the desired asymmetrical PDIs, as illustrated in Scheme 2 (Approach A) [21,37]. Moreover, the partial hydrolysis of PTCDA, which provides a mixed anhydride dicarboxylate salt, is another common approach to access asymmetrical PDIs via successive imidization reactions (Scheme 2, Approach B) [21,35,37,38].
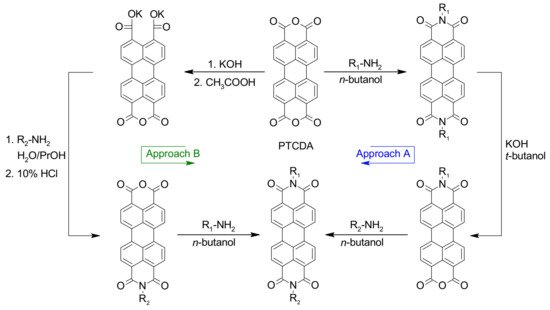
Scheme 2. Synthetic routes for the preparation of asymmetrical PDIs with different substituents on imide positions.
3. Peptide-PDI Bioconjugates
Over the last two decades, amino acids and peptides have been harnessed to control the solubility, optical properties and/or self-assembly of PDI derivatives by taking advantage of highly directional hydrogen-bonding interactions as well as other non-covalent interactions such as ionic and π−π interactions [39]. In addition, polypeptides are known for their biocompatibility, biodegradability and molecular specificity [40], ultimately supporting the use of peptide-based PDI assemblies for different biomedical applications. Chemically, peptides can be considered as linear polymers/oligomers assembled from the condensation of amino acid building blocks [41]. The 20 natural amino acids, as well as countless unnatural amino acids, allow virtually infinite combinations of sequences and offer an unlimited diversity of physicochemical and structural properties of the resulting polypeptide chains [42]. Herein, we present relevant examples of the use of amino acids and short peptide sequences to modulate the self-assembly of PDI derivatives into tailored nanostructures. The self-assembly of PDI–peptide conjugates and the resulting supramolecular morphologies are modulated by a fine balance of complex intermolecular interactions, such as PDI’s π–π stacking interactions and numerous non-covalent interactions involving side chains and the polyamide backbone, as well as by the conditions of the microenvironment, including solvent polarity, solution ionic strength, pH, and temperature [9,11,39].
3.1. Synthesis of Peptide-Conjugated PDIs
Amino acid– and peptide–PDI conjugates are commonly prepared by respectively introducing amino acids and short peptide sequences at one or both imide positions of PDI. Peptides are usually synthesized on solid support, which involves the attachment of the C-terminal amino acid to the polymeric resin by a covalent bond followed by the sequential addition of individual preactivated amino acids, and the final cleavage of the polypeptide from the solid support [43,44]. In contrast to solution synthesis, solid-phase peptide synthesis (SPPS) simplifies dramatically the purification steps between each reaction, thus reducing synthesis time and increasing yield. Consequently, not only does SPPS offer an efficient route towards automation, but also allows the routine access to high-molecular weight polypeptides of up to 50-residues [45]. Due to the insolubility of PTCDA as PDI precursor in common solvents used in SPPS, Kim et al. developed an alternative method to overcome this problem by initially conjugating the PTCDA to amino acids, accessing soluble PDI–amino acid derivatives [46]. Afterwards, the soluble PDI–amino acid derivative can be conjugated to the target peptide sequence that was elongated on the polymeric support by the common SPPS method (Scheme 3) [46].
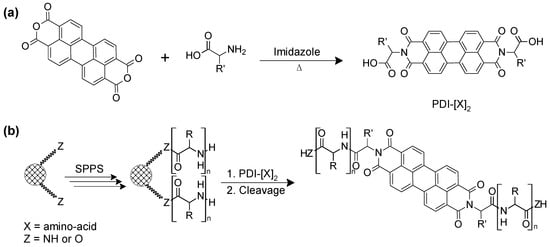
Scheme 3. (a) Synthetic route to amino acid-conjugated PDIs and (b) preparation of peptide-conjugated PDI through the SPPS method followed by their cleavage from the solid support.
3.2. Sequence-Dependent Self-Assembly
Specific variations within peptide sequences have been used to control the self-assembly process of PDI conjugates and to modulate the morphology of the resulting nanostructures through a delicate balance of non-covalent interactions (hydrogen bonding, hydrophobicity, ionic bonding) involving specific residue side chains. Short dipeptides GX (where X = D or Y) were used to enhance the solubility and to modulate the self-assembly properties of PDI in polar organic solvents and aqueous solution [47]. It was observed that, by varying the X residue of the PDI–[GX]2 bola-amphiphile conjugates from hydrophobic Tyr to hydrophilic Asp, the balance between hydrogen bonding and π–π stacking interactions were altered, ultimately affecting the morphology of the assemblies and their optical properties. For instance, in aqueous sodium bicarbonate buffer (pH 10.8), dimethyl sulfoxide (DMSO), dimethylformamide (DMF), tetrahydrofuran (THF), or acetone, the symmetric PDI–[GY]2 formed chiral nanofibers, whereas PDI–[GD]2 assembled into achiral spherical aggregates in buffer and DMSO. In addition, PDI–[GY]2 formed a gel in DMF, while organogels were observed for the PDI–[GD]2 derivative in this polar aprotic solvent. An exhaustive study of structure-assembly relationships revealed how peptide’s physicochemical properties and length, asymmetric substitution at the imide positions, and stereocenter inversion can affect the thermodynamics of the self-assembly of peptide–PDI hybrid molecules [39]. A set of peptide–PDI conjugates were synthetized, all encompassing three units: (i) a glycine residue at the N-terminal position used as a low-steric hindrance linker; (ii) a central variable region composed of three L or D amino acids to evaluate the impact of increasing the peptide hydrophobicity and the role of stereocenters; and (iii) a C-terminal charged region composed of one to three Glu residues to enhance the hydrosolubility and examine the effect of the charge density on the assembly process (Figure 2) [39]. Moreover, to induce a strong amphiphilic character, one of the peptide sequences was replaced by a hydrophobic hexyl chain. It was observed that peptide hydrophobicity and an asymmetrical hexyl substitution induce significant changes on the aggregation thermodynamics of the bioconjugates (Figure 2a,b). In contrast, varying the peptide length, the C-terminal charged region length or the stereocenter inversion induced a significantly lower impact on the aggregation thermodynamics, while having an effect on the peptide-driven self-assembly of PDI nanofibers [39]. Overall, these studies revealed that the physicochemical properties of the residue side chains, the configuration of stereocenters, and the symmetric/asymmetric conjugation can be exploited to dictate the non-covalent interactions that drive the self-assembly of peptide-conjugated PDIs and the final morphology of the assemblies.
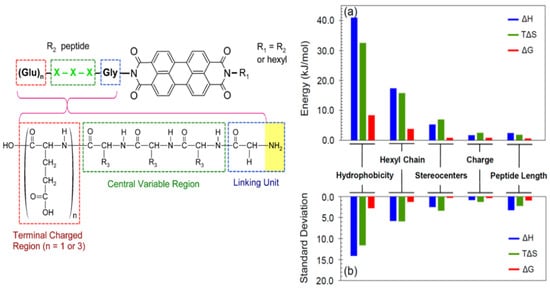
Figure 2. Structure of the peptide–PDI conjugates and impact of structural modulation on the aggregation thermodynamics. The histogram shows (a) the deviation between the lowest and the highest values for each parameter, and (b) the standard deviation as a more collective measure of variability [39]. Copyright © 2014 American Chemical Society.
Inspired by the β-continuous interface of the bovine peroxiredoxin-3 protein, a short heptapeptide (IKHLSVN) was conjugated to PDI in order to control self-assembly into organic semiconductor nanostructures [48]. The designed self-assembling peptide encompassed three different regions: (i) a glycine or ethylamino linker attached to an imide position of the PDI to reduce steric hindrance between the PDI core and the peptide sequence, (ii) a β-sheet-forming peptide, and (iii) a terminal unit composed of glutamic acid residues to assist solubility and to trigger the assembly of peptide–PDI conjugates by pH jump. Two groups of peptide–PDI derivatives were prepared. The first group results from the symmetrical substitution of PDI with peptide sequences, whereas for the second group the PDI core was replaced with perylene imide bis-ester to generate asymmetrical derivatives. Furthermore, for one derivative of each group, the peptide core was attached to the PDI via the amino terminus using a glycine linker, while for the other derivative the peptide core was attached via the carboxyl terminus using an ethylamino linker. The symmetrically substituted PDI showed spectral profiles characteristic of monomers in DMSO. However, UV-visible spectral profiles of bis-ester-functionalized PDI displayed some aggregation, with predominantly monomeric species. In aqueous media, these peptide–PDI conjugates self-assembled into H-aggregate suprastuctures. Particularly, all peptide–PDI derivatives self-assembled into extensive fibril networks in aqueous solution, except for the bis-ester-functionalized PDI for which the peptide was attached via the C-terminal position. This compound formed amorphous, plate-like accretions. Furthermore, reversing the peptide sequence, i.e., N- to C-, for the symmetric derivatives led to the formation of short fibrils and thread-like assemblies instead of ribbon-like structures. This observation highlights the importance of the attachment mode of peptide to the PDI core towards self-assembly and final morphology of the assemblies.
Similarly, the N-(tetra (L-alanine) glycine)-N′-(1-undecyldodecyl) functionalized perylene-3,4,9,10-tetracarboxyl diimide was designed as an asymmetric amphiphilic derivative in order to elucidate how molecular-scale interactions govern the overall self-assembly process [49]. The oligopeptide block on one of the imide nodes of the PDI core provided aggregation directionality through hydrogen bonding and π−π stacking interactions. In chloroform, which was chosen to strengthen inter-peptide hydrogen bonds, this asymmetric amphiphilic PDI derivative adopted a right-handed helical arrangement due to the delicate balance between π–π stacking involving PDI cores and the network of hydrogen bonds between β-sheet-forming peptides. Interestingly, the addition of trifluoroacetic acid (TFA) to the self-assembling media, which was used as a hydrogen-bonding breaking agent, induced the transition of the nanofibers into small aggregates. These aggregates could be brought back into nanofibers by the addition of triethylamine (TEA), which was used to neutralize TFA, favoring the formation of H-bonding between the peptide blocks [49].
Computational simulation and experimental studies have been combined to understand the relationships between the sequence and the self-assembly process of π-conjugated peptides to ultimately predict the resulting supramolecular organisations and photophysical properties from the peptide sequence. For instance, it was shown that increasing the hydrophobicity of the closest residue attached to the PDI core can modulate the photophysical responses in aqueous solution via the conversion of J aggregates, or liquid-crystalline-type materials, to H-type aggregates [50]. In addition, the relationships between the resulting morphologies and the molecular structure of a small library of peptide–PDI derivatives bearing a variable number of L-alanine units as well as methylene, ethylene, and propylene spacers were investigated [51]. It was revealed that the number of L-alanine units in the β-strand peptide segments and the length of the spacer affected the morphology of the resulting suprastructures. In addition, it was shown, through molecular dynamic simulations, that there is a complex interplay between the translation of molecular chirality into supramolecular helicity and the inherent propensity for well-defined one-dimensional aggregation into β-sheet-like superstructures in the presence of a central chromophore [51]. Finally, a symmetric PDI–tripeptide conjugate, which was obtained by introducing a KPA tripeptide block at the 1 and 7 bay positions of PDI via a 2-(2-aminoethoxy)-ethoxyl linker, self-assembled into β-sheet nanohelices directed by hydrogen-bonding [52].These resulting supramolecular structures were particularly sensitive to thermal and ultrasound stimuli. For instance, upon heating/cooling and sonication of the peptide–PDI sample, an interconversion of the supramolecular chirality between left- and right-handed nanostructures was observed [52].
Except for glycine, which is achiral, α-amino acids have an S configuration and are designated as L using the Fischer configurational system [42]. It has been reported that the presence of chiral proximal residues in close proximity to the achiral PDI, for symmetrical peptide–PDI derivatives, influences π–π stacking interactions and induces helical chirality to the PDI core. However, when chiral residues were located distant to the PDI core, or when an isolated stereocenter was introduced in proximal distance of PDI, no effect of chirality was observed during self-assembly [53]. The designed peptide sequences with stereogenic positions and stereochemical configurations included three blocks: (i) an achiral glycine used as a spacer between PDI and the peptide, (ii) three variable residues forming the central block, and (iii) a terminal block of three ionizable glutamic acid residues to assist solubility and pH-triggered aggregation. It was observed that the self-assembly process is modulated by the β-sheet-forming potential of the peptide moieties and the π–π stacking interactions of PDI units. Interestingly, an inversion of the stereocenter within the proximal residues revealed chiral influence. In contrast, an asymmetrical peptide–PDI derivative obtained by the introduction of an alkyl chain at one of the amide nodes, generated an amphiphilic PDI conjugate and disrupted the chiral-mediated self-assembly [53].
Besides, it was observed that the symmetrical conjugation of FF dipeptide to the PDI core leads to a helical assembly due to the chirality of amino acids as well as the co-facial π–π stacking of PDI units [54]. Furthermore, it was shown that there is a close relationship between the translation of molecular chirality into supramolecular helicity and the one-dimensional assembly into well-defined β-sheet-like suprastructures [51]. Overall, these structure–assembly relationship studies have indicated that the morphology of the resulting peptide-conjugated PDI nanostructures can be, to some extent, controlled by modulating the peptide sequence. Moreover, the stereocenters embedded in the peptide backbone can be exploited, under specific conditions, to induce a chiral morphology to the resulting assemblies.
3.3. Solvent-Dependent Self-Assembly
As described above, several substituted water-soluble PDIs have been obtained by functionalizing the PDI core at their imide positions with amino acids and short peptides. Not only do the physicochemical properties of the conjugated moieties dictate the self-assembly behaviour of the PDI core as well as the morphology of the final aggregates, but the solvent also strongly influences the thermodynamics and kinetics of self-assembly [54,55,56]. It is generally assumed that the aggregation constant of PDI derivatives decreases with increasing solvent polarity [57]. The photophysical and aggregation propensities of PDI–[X]2 symmetrical derivatives, where X is a residue with an aromatic group (Y, W or F), were evaluated in various organic solvents [58]. This study revealed that all derivatives self-assemble into amorphous aggregates, excepted for PDI–[Y]2 that forms J-type aggregates in methanol. Although it is still unclear why this effect manifested only in methanol, the authors suggested that the formation of J-aggregates may be possible due to the network of hydrogen bonds involving the hydroxyl groups of tyrosine residues and the carbonyl groups of the PDI core. In pyridine and acetone, PDI–[F]2 and PDI–[Y]2 showed a higher propensity to aggregate than PDI–[W]2, however, the origin of this tendency was unclear. Furthermore, the NMR data indicated a large degree of aggregation in DMSO for all three PDI–peptide derivatives, although the absorption and the fluorescence spectra were both characteristics of soluble and monomeric PDIs [58]. In another study, it was reported that the relatively polar nature of chloroform, in contrast to THF and DMF, facilitates the formation of intermolecular H-bonding of PDI–[F]2, which ultimately leads to J-aggregates [59]. In chloroform, PDI-[F]2 assembled into vesicular suprastructures through the formation of right-handed helix, involving intermolecular H-bonding in lateral and π–π stacking in longitudinal growth directions. As shown in Figure 3, the increase in solvent polarity correlated well with a decrease in the diameter of the vesicles assembled from the PDI–[F]2.

Figure 3. Transmission electron microscopy images of PDI-[F]2 assembled in (a) CHCl3, (b) THF and (c) DMF [59]. Copyright © 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
This entry is adapted from the peer-reviewed paper 10.3390/nano12071223
This entry is offline, you can click here to edit this entry!