Mitophagy, the selective removal of dysfunctional mitochondria by autophagy, is critical for regulating mitochondrial quality control in many physiological processes, including cell development and differentiation. On the other hand, both impaired and excessive mitophagy are involved in the pathogenesis of different ageing-associated diseases such as neurodegeneration, cancer, myocardial injury, liver disease, sarcopenia and diabetes. The best-characterized mitophagy pathway is the PTEN-induced putative kinase 1 (PINK1)/Parkin-dependent pathway. However, other Parkin-independent pathways are also reported to mediate the tethering of mitochondria to the autophagy apparatuses, directly activating mitophagy (mitophagy receptors and other E3 ligases). In addition, the existence of molecular mechanisms other than PINK1-mediated phosphorylation for Parkin activation was proposed. The adenosine50-monophosphate (AMP)-activated protein kinase (AMPK) is emerging as a key player in mitochondrial metabolism and mitophagy.
- mitophagy
- mitochondria
- ubiquitin
- PINK1–Parkin pathway
- Parkin activation
- mitophagy receptors
- E3 ligases
- AMPK
- ULK1
1. Introduction
2. Regulation of Mitophagy
2.1. Chemical and Natural Mitophagy Triggers
In cultured cell lines, it is experimentally advantageous to use chemical reagents to trigger PINK1–Parkin-mediated mitophagy. In this regard, different compounds were described, including many protonophores, inhibitors of the respiratory chain, and novel chemical compounds and naturally occurring substances.
2.2. PINK1–Parkin Axis
The best-known mitochondrial stress signaling pathway is the PINK1–Parkin-driven mitophagy, which mediates the specific ubiquitination and subsequent removal of dysfunctional/damaged mitochondria by recruiting autophagic machinery [46,66].
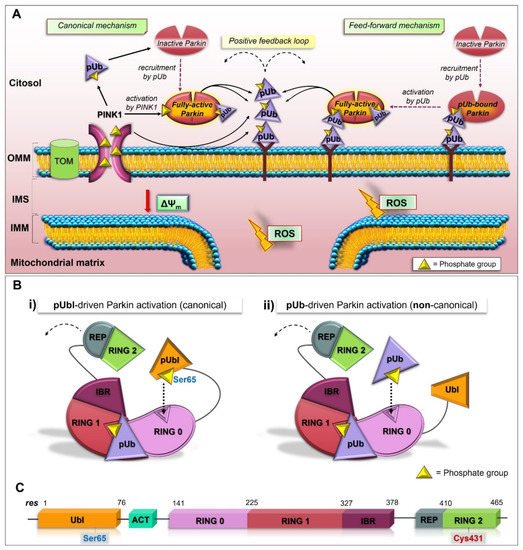
2.3. Structure and Activation of Parkin
PINK1 acts as a mitochondrial damage sensor, and when its import is arrested (under bioenergetics stress, loss in ∆Ψm), it accumulates on the OMM of dysfunctional mitochondria and phosphorylates Ub molecules (pUb). The interaction between pUb and RING1 (at a site consisting of His302 and Arg305) of Parkin leads to the disengagement of the Ubl domain from the core structure, resulting in the conformational rearrangements, which in part liberate Parkin from the inhibitory interactions and promote its accumulation at the surface of mitochondria [76,82,83,84,85,86,87,88,89]. Therefore, the pUb acts as a receptor for Parkin recruitment [90], and the phosphoserine binding on RING1 govern Parkin localization.
2.4. The Feed-Forward Mechanism of Parkin Activation
The addition of new Ub onto the OMM generates a virtuous circle since the presence of more substrates for PINK1 further increases the content of poly-phosphorylated Ub, leading to the extra activation and recruitment of Parkin to mitochondria. This results in a self-amplifying, feed-forward loop that, at the end, directs mitochondria along the mitophagy pathway.
2.5. Molecular Links between PINK1/Parkin-Mediated Mitophagy and Mitochondrial Dynamics
The Parkin-mediated ubiquitination of Mfn1/2 leads to its proteasomal degradation by a p97-dependent extraction mechanism [107,108,109,110]. Thus, Mfn2 degradation leads to the interdiction of fusion and the induction of fission events as well as to the separation of mitochondria–ER contact sites [110]. The Mfn2 localization in the mitochondria–ER contacts and the detection of PINK1 in MAM (mitochondria-associated membrane) also suggest a PINK1/pUb-mediated Parkin recruitment at the mitochondria–ER sites [111]. Different from the autophagy pathway, mitophagy may exhibit an antagonistic and reciprocal relationship with mitochondria–ER contacts as their reduction is functional to the Parkin-mediated ubiquitination of substrates, its recruitment to the OMM and mitochondrial turnover [111].
2.6. Deubiquitinating Enzymes and PTEN-L as Regulators of Mitophagy
The ubiquitination process is reversible and balanced by the activities of deubiquitinating enzymes (DUBs), which modulate protein turnover by removing Ub from ubiquitinated substrates. Many DUBs, such as USP15 (Ub-specific cysteine protease 15), USP30, USP33 and USP35 regulate mitochondrial homeostasis and antagonize PINK1-Parkin-driven mitophagy [92,115,116,117,118]. In this regard, a mechanism of action of USP30-mediated K6-linkage-specific deubiquitination was suggested [119,120].
2.7. pUbl-Independent Mechanism of Parkin Activation, the Unexpected Plasticity of RING0 Binding Site
2.8 PINK1 Processing and Stabilization
3. Functions of Mitophagy Receptors
Under physiological and pathological conditions, mitophagy can occur independently of the presence of Parkin. These pathways rely on the intervention of receptors (BNIP3, NIX, FUNDC1, BCL2L13, FKBP8) constitutively localized in the OMM (via the C-terminal transmembrane (TM) domain) and containing a conserved LIR (LC3-interacting region) motif (at N-terminal region), which allows their association with phagophore on its LC3-decorated membrane.
3.1. Other Promoters of Mitophagy: Cardiolipin and Novel E3 Ligases
3.2. Mitophagosome Synthesis: Redundancy and Positive Feedback Signals, and the Role of mTORC1
3.3. AMPK/ULK1 Axis in Mitophagy Cascade
In response to mitochondrial damage, as well as under energetic stress, the AMPK complex (consisting of a catalytic α, a scaffolding β, and a regulatory γ subunit) acts as a sensor. It engages downstream effectors implicated in metabolic processes, autophagy, and in different aspects of mitochondrial homeostasis, including biogenesis, dynamics, and, ultimately, the clearance of damaged mitochondria, to restore homeostasis. Canonically, a radical increase in the cellular AMP/ATP ratio triggers the full activation of AMPK due to AMP/ADP binding to the γ subunit and the subsequent phosphorylation of the Thr172 by the upstream kinase liver kinase B1 (LKB1) [57]. The direct link of AMPK-mediated, energy-sensing function to the autophagy process is represented by the Ser/Thr kinase ULK1 [219]. In addition to ULK1, AMPK also interacts with ATG9 and components of the Class III PI3K complex 1 of the autophagy pathway [57]. ULK1 represents the most upstream activator of the autophagic pathway and is extensively phosphorylated by AMPK [203,220,221,222]. ULK1 is a core constituent of the autophagy pre-initiation complex with ATG101, ATG13, and FIP200. All of these components are substrates of ULK1 kinase activity as well as ULK1 itself. Additional targets of ULK1 were reported, including AMBRA1, VPS34, syntenin-1, TAB2, Raptor, FUNDC1, BNIP3, ATG14, ATG16L, Sec16a, and Sec23a [165,174,223,224,225,226,227,228,229,230,231,232]. In addition to the autophagy activation, it is becoming more evident that the AMPK/ULK1 axis plays a crucial role in promoting mitophagy [233,234]. Indeed, ULK1- or AMPK-deficient cell lines exhibit an increased accumulation of morphologically altered mitochondria, suggesting that the AMPK-dependent phosphorylation of ULK1 is crucial for the selective clearance of mitochondria [220,235,236]. In this regard, AMPK-mediated ULK1 phosphorylation at Ser555 regulates ULK1 translocation to mitochondria and mitophagy [234,237]. As the proton ionophore CCCP is a strong inducer of PINK1–Parkin-mediated mitophagy and AMPK by ATP depletion, new studies are developing in order to establish which relationship links PINK/Parkin pathway with the AMPK/ULK1 axis. In this regard, exactly how Parkin first senses problems with mitochondria and how specific phosphorylation events orient towards mitophagy remain to be defined [238]. Another important question is how AMPK regulates mitophagy in skeletal muscle. In this regard, new PINK1–Parkin-independent mechanisms and the activation of specific, subcellular AMPK pools were reported [239,240,241].
3.4. The Early Role of the AMPK/ULK1 Axis in Triggering the Rapid Activation of Parkin
As described above, although it is well known that Parkin can sense mitochondrial stress and promote mitophagy, the initial input in dictating the earliest Parkin recruitment remains to be clarified. In this sense, a model revealing the implication of the AMPK-ULK1 axis in starting the first translocation of Parkin onto the OMM was recently proposed in vivo and in vitro (Figure 2A) [238]. In particular, Parkin was reported as a novel ULK1 substrate. Under mitochondrial stress (exposure to CCCP or valinomycin), the immediate activation of cytosolic AMPK (within 2 min) leads to the phosphorylation of its downstream substrates, including ULK Ser555, Raptor Ser 792, MFF Ser146, and ACC Ser79. At the same time, ULK1 activation results in the specific phosphorylation of Parkin Ser108 (P-Parkin-108), an event that seems to be localized in the cytoplasmic region [238]. This phosphorylation falls in a new highly conserved region named the ACT element (activating element). This short region is localized in the flexible linker between the UBL and RING0 domains and is suggested to be critical for Parkin activation (Figure 2A, Top) [79]. Interestingly, phosphoproteomic analyses previously revealed the phosphorylation of Parkin Ser108 in brown fat, although the kinase responsible for this event was not identified identified [242]. In parallel to P-Parkin108, phosphorylations of Beclin Ser30 and ATG16L1 Ser278, two ULK1 substrates [224,230], were also observed. Within 10 min of CCCP exposure, the recruitment of AMPK and ULK1 to mitochondria leads to MFF phosphorylation and mitochondrial fission. Only at later time points (after 30 min) does the phosphorylation of Parkin Ser65 occur. This event coincides with the ubiquitination of the substrates of Parkin, CISD1 and Mfn2, and TBK activation at Ser172 [238]. This study demonstrates that the rapid and greatest PINK1-mediated phosphorylation of Parkin Ser65 requires the ULK1-dependent phosphorylation of Parkin at Ser108. In addition, these findings highlight the crucial and early role played by the AMPK/ULK1 axis in mitophagy and place Parkin regulation downstream of AMPK/ULK1, revealing a new route to modulating Parkin.
3.5. Other Scenarios of AMPK- and ULK1-Mediated Mitophagy
3.6. The Mitochondrial Pool of AMPK (mitoAMPK) Governs the Spatial Specificity of Energetic Stress-Induced Mitophagy
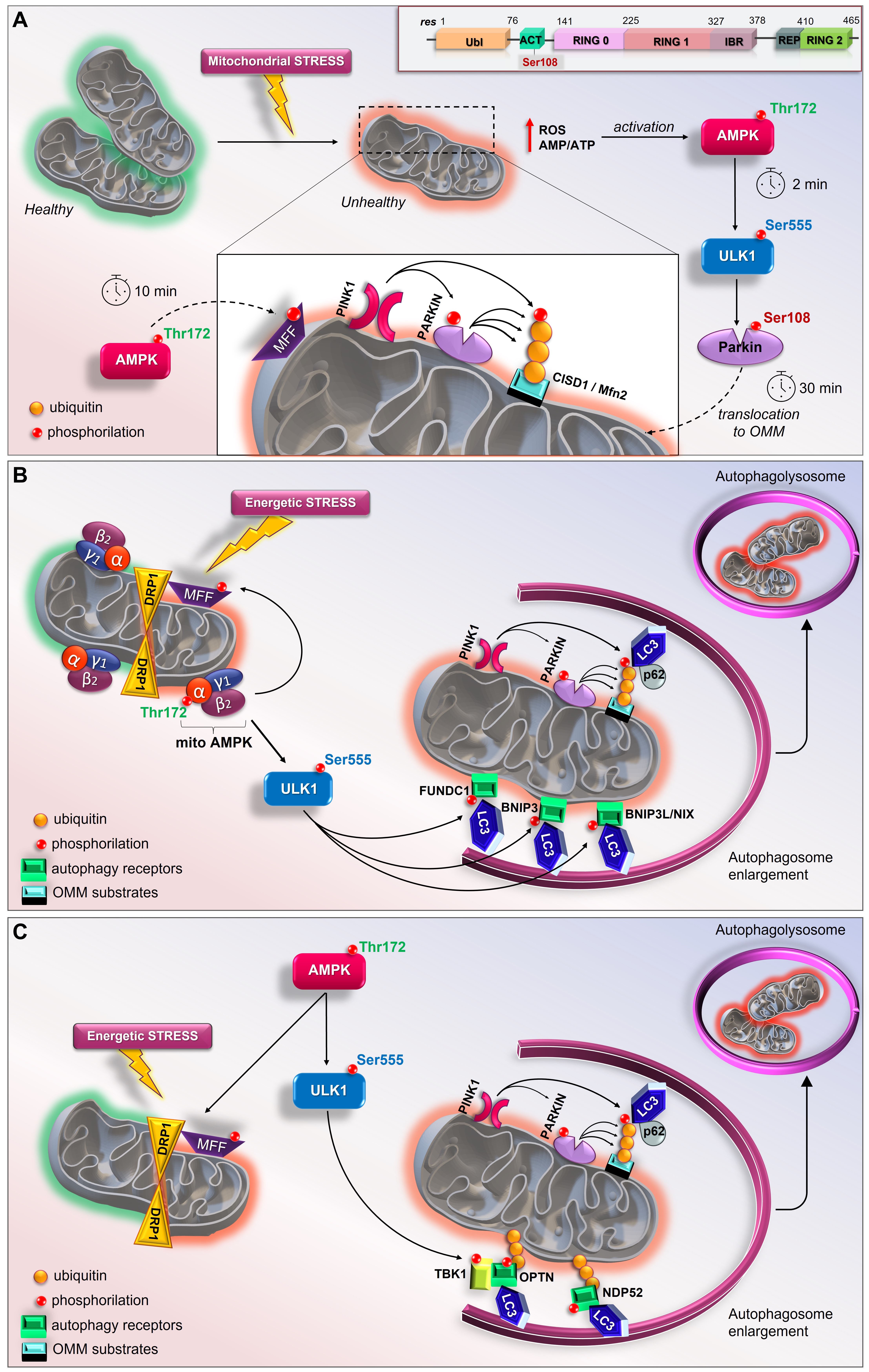
Figure 2. New current models of AMPK/ULK1-mediated mitophagy. (A) Top, schematic representation of primary structure of Parkin showing ACT element with Ser108 residue. In mouse livers and primary hepatocytes AMP/ULK1 axis triggers the rapid activation of Parkin (by Hung et al.) [238]. Upon mitochondrial depolarization, AMP/ATP imbalance and increases in mtROS immediately activate AMPK/ULK1 signaling. Therefore, in the cytoplasm ULK1 phosphorylates Parkin at Ser108 within 2 min of depolarization treatment. Later, AMPK phosphorylates MFF to induce mitochondrial fission and PINK1 phosphorylates Parkin at Ser65; (B) In skeletal muscle, mitoAMPK regulates the spatial specificity of mitophagy in a context of mitochondrial remodeling (by Drake et al.) [241]. Following energetic stress, mitoAMPK is activated in vivo and promotes ULK1-mediated formation of autophagosome which fuse with lysosomes to allow the complete mitochondrial degradation; (C) In C2C12 myotubes, AMPK coordinates mitophagy through mitochondrial fission via MFF and TBK1-mediated autophagosomal engulfment via ULK1 activation (by Seabright et al.) [239].
This entry is adapted from the peer-reviewed paper 10.3390/cells11010030