Mitochondria are semi-autonomous intracellular double membrane-bound organelles, which include an outer membrane, a highly folded inner membrane (crista), a matrix space surrounded by the inner membrane, and an inter-membrane space between the inner and outer membranes. Usually, a cell has hundreds or thousands of mitochondria, which can occupy up to 25% of the cellular cytoplasm. Mitochondria are a convergence point for glucose, glutamine, and lipid metabolism. The primary function of mitochondria is to support the TCA cycle and aerobic respiration by oxidative phosphorylation, generating ATP through the mitochondrial respiratory chain to fulfill the energy needs for cell survival. One unique feature of mitochondria is that they possess their own supercoiled, double-stranded circular genetic material called mitochondrial DNA (mtDNA) that encodes rRNAs, tRNAs, and proteins essential for electron transport and oxidative phosphorylation, as well as their own genetic repair mechanisms. Mitochondrial biogenesis requires the coordinated expression of both mtDNA- and nuclear DNA-encoded genes. Thirteen proteins are encoded by mtDNA, while approximately 1000 mitochondrial proteins are encoded by the nuclear genome, translated in the cytoplasm and transported into the mitochondria by a specific transport system. These two pools of proteins are required to maintain mitochondria as a cellular power hub and a signaling nexus that are essential for normal cell function. Defects in many of the mitochondrial components are causal for a multitude of cellular diseases. Of note, the reprogramming of cellular metabolism and the aberrant redox status have been heralded as major emerging hallmarks of neoplastic transformation. Overall, mitochondrial dysfunction caused by mtDNA mutations, malfunctioned TCA cycle enzymes, electron respiratory chain leakage and subsequent oxidative stress, and/or aberrant oncogenic and tumor suppressor signaling is known to alter cellular metabolic pathways, disrupt redox balance, and cause resistance to apoptosis and therapies that significantly contribute to the development of multiple types of human cancers. In the following sections, we will present current knowledge on these aspects of mitochondrial dysfunction pertaining to the pathologies of various forms of human malignancies.
- mitochondria
- dysfunction
- cancers
- TCA cycle
- electron transport chain
- oxidative phosphorylation
- oncogene
- tumor suppressor
1. Mitochondrial TCA Cycle and Human Cancer
Glucose represents a key component of the daily diet and can be actively transported into cells and converted into pyruvate by glycolysis in the cytosol. When oxygen is limited, pyruvate is converted into lactate by lactate dehydrogenase (LDH). In the presence of oxygen, pyruvate can be transported into mitochondria and decarboxylated to produce acetyl CoA by the pyruvate dehydrogenase complex (PDH) in the mitochondria. The oxidation of acetyl CoA is achieved by the tricarboxylic acid (TCA) cycle, which involves a set of metabolic reactions catalyzed by specific enzymes including citrate synthase, aconitase, isocitrate dehydrogenase (IDH), α-ketoglutarate (α-KG) dehydrogenase complex, succinyl-CoA synthase, succinate dehydrogenase (SDH), fumarate hydratase (FH), and malate dehydrogenase in the mitochondria to generate CO2, H2O, and the bioenergetic products GTP, NADH, and FADH2 (Figure 1) [1][2]. The TCA cycle represents the final converging route for the oxidation of lipids, carbohydrates, and amino acids [3]. Recent studies have shown that mutations in several enzymes involved in the TCA cycle can induce various human cancers.
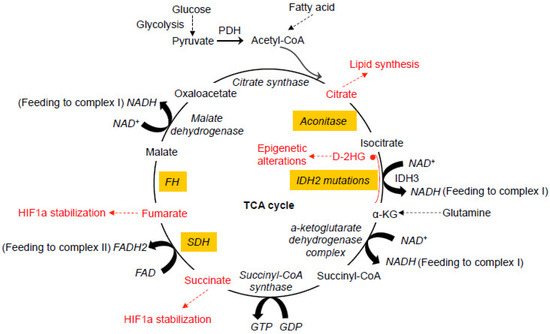
Figure 1. Dysfunctional tricarboxylic acid (TCA) cycle enzymes in cancers. The normal TCA cycle deemed for the breakdown of acetyl-CoA is subject to disruption by oxidative stress and dysfunction of the TCA cycle enzymes, contributing to various cancers. Mutations in IDH2 can lead to the production of D-2HG from α-KG. Abnormal accumulation of 2-HG causes epigenetic landscape alterations and inhibits SDH, resulting in the accumulation of succinyl-CoA and mitochondrial respiration impairment. Mutations in SDH cause abnormal accumulation of succinate and subsequent inhibition of PHDs, resulting in HIF1a stabilization. Like SDH, mutations in FH also cause HIF1a stabilization. Oxidative stress induced by the dysfunction of mitochondrial electron transport chain causes abnormal modification of the iron-sulfur center in aconitase, leading to the exportation of citrate from mitochondria for cholesterol and fatty acid synthesis. All of these abnormal alterations in the TCA cycles have been more or less implicated in neoplastic transformation of different types at different degrees. PDH, pyruvate dehydrogenase. PHD, HIF prolyl hydroxylase. D-2HG, D enantiomer of 2-hydroxyglutarate. α-KG, alpha-ketoglutarate. IDH, isocitrate dehydrogenase. SDH, succinate dehydrogenase. FH, fumarate hydratase.
2. Mitochondrial DNA Mutations and Respiratory Chain Dysfunction in Cancer
2.1. Mitochondrial DNA Mutations in Cancer
Mutations in mtDNA have been found in a spectrum of human cancers [4][5][6] . They can occur at the D-loop region, the protein-coding region, the rRNA genes, and the tRNA genes [7][8][9]. The most common mtDNA alterations in cancers are point mutations and reduced copy numbers, but frame-shifting insertions and large-scale deletions have also been identified [7]. The D-loop region of mtDNA is regarded as the most frequent site of somatic mutations in many cancers. Because mtDNA replication starts at the D-loop, mutations in the D-loop region can affect mtDNA copy number in cancers. In a study investigating mtDNA mutations in 31 gastric cancer patients, 51.6% of mtDNA mutations occurred in the D-loop region, 22.6% in the protein-coding region, and 4% in tRNA genes [10]. Through sequencing analysis of the entire mtDNA from 58 breast cancer samples with paired non-tumorous breast tissues, researchers identified that 52.5% of mtDNA mutations were located in the D-loop region, 37.5% in protein-coding region, and 5% each in the rRNA and tRNA genes. These somatic mutations are thought to cause mitochondrial dysfunction by altering the functions of 12S rRNA (T1499C), 16S rRNA (G1913A), tRNATrp (G5522A), tRNACys (G5809A), as well as ND2 (G5112A), ND4L (G10599A), ND5 (A13878G) of complex I, cytochrome b (T15416C) of complex III, and COI (G6384A, G6768A) and COIII (G9412A, G9774A, A9901C) of complex IV [9]. In hepatocellular carcinoma, somatic mtDNA mutations that affect the functions of tRNAVal (T1659C), tRNAAla (G5650A), ND1 (G3842A), ND4 (11032delA, A11708G), ND5 (12418insA), COI (T6787C), COII (G7976A), and COIII (A9263G, G9267A) were also observed [8]. By sequencing the mitochondrial genomes of 384 prostate cancer patients, researchers found that 15.4% of the tumors harbor mutations in the non-coding control region of mitochondrial genome [11], while ND5 was the most frequently mutated gene in the protein-coding region [11]. Furthermore, sequence analyses of 226 paired tumor and normal tissue samples in five cancers by The Cancer Genome Atlas (TCGA) revealed that the frequencies of deleterious tumor-specific somatic mtDNA mutations are 63% in rectal adenocarcinomas, 53% in colon adenocarcinomas, 36% on ovarian serous cyst adenocarcinomas, 30% in acute myeloid leukemia, and 13% in glioblastoma, highlighting the prevalence of mtDNA mutations in diverse types of cancers. These mtDNA mutations are predicted to impact the functions of encoded proteins that confer a selective advantage in oncogenesis [12]. Some mutation hotspots have been observed in several cancers. The 12418insA mutation, with an adenosine nucleotide insertion in a poly-A sequence at nucleotide position 12,418–12,425 of mtDNA, causes a frame-shift and premature termination of the ND5 gene and results in a truncated polypeptide. This mutation was observed in colorectal cancer, breast cancer, gastric cancer, and hepatocellular cancer and is correlated with defective mitochondrial respiration, higher levels of lactate production, and increased tumorigenesis [8][9][10][13]. By using a cybrid cell model, a heteroplasmic 12418insA mutation was shown to increase ROS generation and decrease oxidative phosphorylation in human cancer cells, as well as promote tumor growth in nude mice [14], suggesting that this hotspot mutation plays a critical role in tumorigenesis. Of note, the G-to-A and T-to-C transitions, which are typical changes that occur upon exposure to free radicals, are rather common, suggesting that these mutations are associated with oxidative stress. In a mtDNA analysis of colon cancer patients, it was found that 70% of the identified mtDNA mutations were T-to-C and G-to-A replacements [15]. Similarly, the G-to-A replacements in the ATP6 gene at positions 8557, 8697, and 8854 of mtDNA were found in breast cancer patients [16]. The accumulation of mtDNA mutations was found to correlate with the degree of malignancy [17]. Furthermore, mutations in mtDNA exacerbate ROS production and oxidative stress [18] to promote more mtDNA mutations, which, in turn, create a vicious cycle critical for tumorigenesis.
2.2. The Complexity in the Roles of mtDNA Mutations in Tumorigenesis
Although the majority of studies suggested a positive role of mtDNA mutations in neoplastic progression, it should be noted that a clear link between mtDNA mutations and tumorigenesis has yet to be established, in part owing to the hurdle in our ability to directly manipulate mtDNA and the complexity of mutational landscape obtained from different experimental models and tumor samples. It thus remains a gap in knowledge on mtDNA mutations and their biological significance in tumor formation, progression, and metastasis. In some preclinical cellular and mouse xenograft models, depletion of mtDNA in tumor cells was shown to decrease tumorigenic phenotypes or potential [19], while the acquisition of normal mtDNA, e.g., via horizontal transfer of the whole mtDNA from surrounding cells, could restore functional mitochondrial respiration and tumorigenic potential of mtDNA-depleted cancer cells [20][21][22]. These studies indicate that mitochondrial respiration or oxidative phosphorylation may be required for tumorigenic potential under certain conditions. Some mtDNA mutations can be found in both normal and cancer tissues. The deletion of mtDNA at 4977-bp has been implicated in the contribution to malignant transformation in breast cancer patients; however, it was also observed in the benign tissues and surrounding normal tissues [23]. Furthermore, analysis of random point mutations in the mtDNA of cancerous and healthy tissues from 21 colorectal cancer patients revealed that the frequency of mtDNA mutations decreased in colorectal tumors relative to the adjacent healthy tissues [24], suggesting a mutation selection during tumor development and that not all mtDNA mutations contribute to cancer development—some may suppress tumor growth [24]. Using different transmitochondrial cytoplasmic cybrid cell models harboring a panel of pathogenic mtDNA mutations that disrupt the respiratory chain, studies showed that defects in the respiratory chain could either promote or inhibit cell death, depending on the specific alterations in the electron flow and cytosolic cues (e.g., ER stress) [25].
Oncocytoma is a rare, predominantly benign neoplasm characterized by the dramatic accumulation of defective mitochondria that have mtDNA mutations in the control region and protein-encoding region that induce respiration chain defects [26]. The common mtDNA gene mutations in oncocytomas include COI, COII, COIII, ND4, ND5, and CYTB. It has been a puzzle about what restricts oncocytomas to remain a benign disease despite such a spectrum of mutations. A recent study suggests that these genetic defects in mitochondrial respiration block Golgi to lysosome trafficking and autophagy, and activate AMPK and p53, thus limiting tumor growth to only a benign state [27]. The type 2 oncocytomas are closely related to the eosinophilic subtype of chromophobe renal cell carcinoma (ChRCC), as they share similarities in the mutational landscape and transcriptome profile, with ChRCC having acquired additional driver mutations in p53 and PTEN and further genetic instability [27]. This study indicates that the impaired mitochondrial function may be a barrier to tumorigenesis by activating p53 in oncocytomas, while subsequent alterations of p53 and other nuclear genes lead to malignant ChRCC.
In brief, although the majority of evidence supports a role of mtDNA mutations in tumorigenesis and malignant progression, the battle to determine whether an mtDNA mutation is a driver mutation for cancer initiation and progression or merely a passenger mutation that does not determine the development of cancer is predicted to continue. The biological impact of a given mtDNA mutation may vary, depending on the nature of the mutation, the proportion of the mutation in the cell, the effects of the mutation to the respiratory chain, and the interplay of the mutant mtDNA-directed events with cytoplasmic signal pathways. Innovation in mtDNA manipulation in well-defined model systems is essential for clarifying the complexity.
3. Mitochondrial Oxidative Stress and Cancer
The process of mitochondrial oxidative phosphorylation consumes more than 90% of oxygen. Although the most portion of oxygen is fully reduced to water by cytochrome c oxidase, 1–2% is incompletely reduced to superoxide radicals mainly in complex I and III of the respiratory chain [28][29][30][31]. Superoxide in the mitochondrial matrix can be rapidly dismuted by mitochondrial superoxide dismutase 2 (SOD2) yielding H2O2, which can be released into the intermembrane space and cytosol leading to the generation of cytoplasmic ROS. In the presence of a reduced transition metal, H2O2 can be converted into the highly reactive hydroxyl radical OH· [32][33][34][35]. The unpaired electrons are highly reactive, producing chemical modifications that damage proteins, lipids, and nucleotides [36]. Under physiological conditions, free radicals and their derivatives exist in living tissues at low but measurable concentrations, which are determined by the balance between the rate of radical production and the rate of clearance. Free radicals can be eliminated by the reducing equivalent GSH, an abundant cellular thiol and a major determinant of cellular redox equilibrium [37]. Other antioxidant enzymes such as glutathione peroxidase (GPX), glutathione reductase (GR), peroxiredoxin (PRX), thioredoxin, and thioredoxin reductase also play important roles in the clearance of free radicals and the maintenance of redox homeostasis [38].
Mitochondrial respiratory chain dysfunction induced by mtDNA mutations can cause an increase in electron leakage, resulting in an elevation of free radical generation as widely observed in cancer cells [39]. In most cells, mitochondrial complex I and complex III, as well as membrane-bound NADPH oxidases (NOXs) are the main sources of cellular ROS [40][41][42][43]. Interestingly, mitochondrial respiratory chain dysfunction can induce NOX activation [44], suggesting that NOX can be activated in response to mitochondrial dysfunction, contributing to heightened levels of cellular ROS. The escalated ROS generation in cancer cells serves as a messenger to stimulate cell proliferation and as an endogenous source of DNA-damaging agents to promote genetic instability and the development of cancer [45]. DNA contains a large number of ROS-reactive sites [46]. Oxidative damages to DNA may be in the form of base modifications (such as 8-oxoguanine), abasic sites, strand breaks, or various other types of lesions [47]. Of note, ROS production in mitochondria renders mtDNA more susceptible to damage and mutagenesis than the nuclear genome. This is because mtDNA lacks histone protection, has limited DNA damage repair capacity, and is in close proximity to the electron transport chain. As mitochondrial transcription is polycistronic, deletion or insertion of a nucleotide may readily cause the downstream codon frameshifting, resulting in an inability to encode functional products [48]. It was reported that the rate of mtDNA mutation is 10–20 times greater than that of nuclear DNA [49]. Thus, ROS-induced mtDNA mutations and the high susceptibility of mtDNA to mutations play key roles in many diseases, including cancer, aging, and neurodegenerative diseases.
Notably, some key enzymes for energy metabolism can be directly targeted by free radicals. The Fe-S clusters are essential components of the redox-active enzymes within both the TCA cycle and the respiratory chain [50]. Excess superoxide and hydroxyl radicals are capable of reacting with the Fe-S clusters in NADH dehydrogenase, SDH, aconitase, and other enzymes, resulting in their inactivation and subsequent inhibition of production of biological intermediates and energy [51]. NADH dehydrogenase is one of the key enzymes involved in the oxidative phosphorylation, and aconitase is the first rate-limiting enzyme converting citrate into isocitrate in the TCA cycle generating high energy reducing equivalent NADH. Aconitase has been found to be significantly reduced in tumor cells [52]. The impaired activity of aconitase causes a truncation to the TCA cycle, leading to the exportation of considerably large amounts of citrate [53], an essential precursor for the de novo biosynthesis of cholesterol and fatty acid.
4. Summary
Cancer cells exhibit an altered redox status and metabolism, which are associated with mitochondria as they are the major sites of ROS generation and energy metabolism. In this review, we have discussed mitochondrial dysfunction induced by the alterations of the mitochondrial genome, the TCA cycle enzymes, mitochondrial electron transport chain, and the associated oxidative stress. We have also discussed the aberrant oncogene and tumor suppressor signaling in the induction of mitochondrial dysfunction in cancers. Despite the prevalence of mtDNA mutations in different malignancies, their causal roles in cancer development remain incompletely understood and many questions still remain. It is imperative for us to gain a better understanding of why certain mutations appear more commonly in some cancers than in others. This may be in part due to the fact that different types of cancers have distinct underlying drivers and/or specific requirements in the path toward the malignant transformation. It has been known that oncogenic KRAS is the critical driver for the initiation and development of pancreatic cancers, while loss of tumor suppressors, such as PTEN and TP53, are major drivers of prostate cancer development. Mitochondrial dysfunction may be induced under either of these conditions but through distinct mechanisms. Certain control regions of mitochondrial single-nucleotide variants were recently found to co-occur with a gain of Myc oncogene in prostate cancer patients [11]. Different mtDNA mutations may impose divergent influences at varying degrees on the respiratory chain, with some mutations causing critical alterations of the respiratory chain and leading to transformation-promoting oxidative stress, while others may play lesser roles. The alterations of the electron transport chain affect not only electron transport, oxygen consumption, and ATP generation but also cellular redox status, metabolism, and apoptosis. It should be noted that a severe level of mitochondrial dysfunction may cause cell death and thus inhibit tumorigenesis, while mild mitochondrial dysfunction may enhance mitochondrial ROS generation and redox rebalance to stimulate cancer cell proliferation and invasiveness. Thus, a better understanding of the roles of mitochondrial dysfunction in cancer is essential for the future design of effective therapeutic strategies against diverse types of cancers.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21165598
References
- Krebs, H.A. The history of the tricarboxylic acid cycle. Perspect. Biol. Med. 1970, 14, 154–170.
- Krebs, H.A. Rate control of the tricarboxylic acid cycle. Adv. Enzyme Regul. 1970, 8, 335–353.
- Akram, M. Citric acid cycle and role of its intermediates in metabolism. Cell Biochem. Biophys. 2014, 68, 475–478, doi:10.1007/s12013-013-9750-1.
- Modica-Napolitano, J.S.; Singh, K.K. Mitochondrial dysfunction in cancer. Mitochondrion 2004, 4, 755–762.
- Verma, M.; Kagan, J.; Sidransky, D.; Srivastava, S. Proteomic analysis of cancer-cell mitochondria. Nat. Rev. 2003, 3, 789–795.
- Hsu, C.C.; Tseng, L.M.; Lee, H.C. Role of mitochondrial dysfunction in cancer progression. Exp. Biol. Med. 2016, 241, 1281–1295, doi:10.1177/1535370216641787.
- Lee, H.C.; Huang, K.H.; Yeh, T.S.; Chi, C.W. Somatic alterations in mitochondrial DNA and mitochondrial dysfunction in gastric cancer progression. World J. Gastroenterol. 2014, 20, 3950–3959, doi:10.3748/wjg.v20.i14.3950.
- Yin, P.H.; Wu, C.C.; Lin, J.C.; Chi, C.W.; Wei, Y.H; Lee, H.C. Somatic mutations of mitochondrial genome in hepatocellular carcinoma. Mitochondrion 2010, 10, 174–182, doi:10.1016/j.mito.2009.12.147.
- Tseng, L.M.; Yin, P.H.; Yang, C.W.; Tsai, Y.F.; Hsu, C.Y.; Chi, C.W.; Lee, H.C. Somatic mutations of the mitochondrial genome in human breast cancers. Genes Chromosom. Cancer 2011, 50, 800–811, doi:10.1002/gcc.20901.
- Hung, W.Y.; Wu, C.W.; Yin, P.H.; Chang, C.J.; Li, A.F.; Chi, C.W.; Wei, Y.H.; Lee, H.C. Somatic mutations in mitochondrial genome and their potential roles in the progression of human gastric cancer. Biochim. Biophys. Acta 2010, 1800, 264–270, doi:10.1016/j.bbagen.2009.06.006.
- Hopkins, J.F.; Sabelnykova, V.Y.; Weischenfeldt, J.; Simon, R.; Aguiar, J.A.; Alkallas, R.; Heisler, L.E.; Zhang, J.; Watson, J.D.; Chua, M.; et al. Mitochondrial mutations drive prostate cancer aggression. Nat. Commun 2017, 8, 656, doi:10.1038/s41467-017-00377-y.
- Larman, T.C.; DePalma, S.R.; Hadjipanayis, A.G.; Cancer Genome Atlas Research N, Protopopov, A.; Zhang, J.; Gabriel, S.B.; Chin, L.; Seidman, C.E.; Kucherlapati, R.; et al. Spectrum of somatic mitochondrial mutations in five cancers. Proc. Natl. Acad. Sci. USA 2012, 109, 14087–14091, doi:10.1073/pnas.1211502109.
- Polyak, K.; Li, Y.; Zhu, H.; Lengauer, C.; Willson, J.K.; Markowitz, S.D.; Trush, M.A.; Kinzler, K.W.; Vogelstein, B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat. Genet. 1998, 20, 291–293, doi:10.1038/3108.
- Park, J.S.; Sharma, L.K.; Li, H.; Xiang, R.; Holstein, D.; Wu, J.; Lechleiter, J.; Naylor, S.L.; Deng, J.J.; Lu, J.; et al. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum. Mol. Genet. 2009, 18, 1578–1589, doi:10.1093/hmg/ddp069.
- Habano, W.; Nakamura, S.; Sugai, T. Microsatellite instability in the mitochondrial DNA of colorectal carcinomas: Evidence for mismatch repair systems in mitochondrial genome. Oncogene 1998, 17, 1931–1937, doi:10.1038/sj.onc.1202112.
- Grzybowska-Szatkowska, L.; Slaska, B.; Rzymowska, J.; Brzozowska, A.; Florianczyk, B. Novel mitochondrial mutations in the ATP6 and ATP8 genes in patients with breast cancer. Mol. Med. Rep. 2014, 10, 1772–1778, doi:10.3892/mmr.2014.2471.
- Nishikawa, M.; Nishiguchi, S.; Shiomi, S.; Tamori, A.; Koh, N.; Takeda, T.; Kubo, S.; Hirohashi, K.; Kinoshita, H.; Sato. E.; et al. Somatic mutation of mitochondrial DNA in cancerous and noncancerous liver tissue in individuals with hepatocellular carcinoma. Cancer Res. 2001, 61, 1843–1845.
- Hahn, A.; Zuryn, S. Mitochondrial Genome (mtDNA) Mutations that Generate Reactive Oxygen Species. Antioxidants 2019, 8, doi:10.3390/antiox8090392.
- Cavalli, L.R.; Varella-Garcia, M.; Liang, B.C. Diminished tumorigenic phenotype after depletion of mitochondrial DNA. Cell Growth Differ. 1997, 8, 1189–1198.
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94, doi:10.1016/j.cmet.2014.12.003.
- Dong, L.F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. Elife 2017, 6, doi:10.7554/eLife.22187.
- Bajzikova, M.; Kovarova, J.; Coelho, A.R.; Boukalova, S.; Oh, S.; Rohlenova, K.; Svec, D.; Hubackova, S.; Endaya, B.; Judasova, K. Reactivation of Dihydroorotate Dehydrogenase-Driven Pyrimidine Biosynthesis Restores Tumor Growth of Respiration-Deficient Cancer Cells. Cell Metab. 2019, 29, 399–416, doi:10.1016/j.cmet.2018.10.014.
- Ye, C.; Shu, X.O.; Wen, W.; Pierce, L.; Courtney, R.; Gao, Y.T.; Zheng, W.; Cai, Q. Quantitative analysis of mitochondrial DNA 4977-bp deletion in sporadic breast cancer and benign breast diseases. Breast Cancer Res. Treat. 2008, 108, 427–434, doi:10.1007/s10549-007-9613-9.
- Ericson, N.G.; et al. Decreased mitochondrial DNA mutagenesis in human colorectal cancer. PLoS Genet. 2012, 8, e1002689, doi:10.1371/journal.pgen.1002689.
- Kwong, J.Q.; Henning, M.S.; Starkov, A.A.; Manfredi, G. The mitochondrial respiratory chain is a modulator of apoptosis. J. Cell Biol. 2007, 179, 1163–1177, doi:10.1083/jcb.200704059.
- Gasparre, G.; Romeo, G.; Rugolo, M.; Porcelli, A.M. Learning from oncocytic tumors: Why choose inefficient mitochondria? Biochim. Biophys. Acta 2011, 1807, 633–642, doi:10.1016/j.bbabio.2010.08.006.
- Joshi, S.; Tolkunov, D.; Aviv, H.; Hakimi, A.; Yao, M.; Hsieh, J.J; Ganesan, S.; Chan, C.S.; White, E. The Genomic Landscape of Renal Oncocytoma Identifies a Metabolic Barrier to Tumorigenesis. Cell Rep. 2015, 13, 1895–1908, doi:10.1016/j.celrep.2015.10.059.
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605.
- Scandalios, J.G. The rise of ROS. Trends Biochem. Sci. 2002, 27, 483–486.
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344.
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000, 25, 502–508.
- Le Bras, M.; Clement, M.V.; Pervaiz, S.; Brenner, C. Reactive oxygen species and the mitochondrial signaling pathway of cell death. Histol. Histopathol. 2005, 20, 205–219, doi:10.14670/HH-20.205.
- Szibor, M.; Richter, C.; Ghafourifar, P. Redox control of mitochondrial functions. Antioxid. Redox Signal. 2001, 3, 515–523, doi:10.1089/15230860152409149.
- Mannick, J.B.; Schonhoff, C.; Papeta, N.; Ghafourifar, P.; Szibor, M.; Fang, K.; Gaston, B. S-Nitrosylation of mitochondrial caspases. J. Cell Biol. 2001, 154, 1111–1116.
- Goettsch, W.; Lattmann, T.; Amann, K.; Szibor, M.; Morawietz, H.; Munter, K.; Muller, S.P.; Shaw, S.; Barton, M. Increased expression of endothelin-1 and inducible nitric oxide synthase isoform II in aging arteries in vivo: Implications for atherosclerosis. Biochem. Biophys. Res. Commun. 2001, 280, 908–913.
- Khan, S.R. Hyperoxaluria-induced oxidative stress and antioxidants for renal protection. Urol. Res. 2005, 33, 349–357.
- Hall, A.G. Review: The role of glutathione in the regulation of apoptosis. Eur. J. Clin. Invest. 1999, 29, 238–245.
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214.
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updates 2004, 7, 97–110.
- Drose, S.; Brandt, U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 2012, 748, 145–169, doi:10.1007/978-1-4614-3573-0_6.
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13, doi:10.1042/BJ20081386.
- Bleier, L.; Drose, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta 2013, 1827, 1320–1331, doi:10.1016/j.bbabio.2012.12.002.
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313, doi:10.1152/physrev.00044.2005.
- Dikalov, S. Cross talk between mitochondria and NADPH oxidase. Free Radic Biol Med. 2011, 51, 1289-1301.
- Zhou, Y.; Hileman, E.O.; Plunkett, W.; Keating, M.J.; Huang, P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood 2003, 101, 4098–4104.
- Baynes, J.W. The Maillard hypothesis on aging: Time to focus on DNA. Ann. N. Y. Acad. Sci. 2002, 959, 360–367.
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285.
- Zastawny, T.H.; Dabrowska, M.; Jaskolski, T.; Klimarczyk, M.; Kulinski, L.; Koszela, A.; Szczesniewicz, M.; Sliwinska, M.; Witkowski, P.; Olinski, R. Comparison of oxidative base damage in mitochondrial and nuclear DNA. Free Radic. Biol. Med. 1998, 24, 722–725.
- Olgun, A.; Akman, S.; Serdar, M.A.; Kutluay, T. Oxidative phosphorylation enzyme complexes in caloric restriction. Exp. Gerontol. 2002, 37, 639–645.
- Lill, R.; Muhlenhoff, U. Iron-sulfur-protein biogenesis in eukaryotes. Trends Biochem. Sci. 2005, 30, 133–141, doi:10.1016/j.tibs.2005.01.006.
- Gardner, P.R.; Nguyen, D.D.; White, C.W. Aconitase is a sensitive and critical target of oxygen poisoning in cultured mammalian cells and in rat lungs. Proc. Natl. Acad. Sci. USA 1994, 91, 12248–12252, doi:10.1073/pnas.91.25.12248.
- Hernanz, A.; de la Fuente, M. Characterization of aconitate hydratase from mitochondria and cytoplasm of ascites tumor cells. Biochem. Cell Biol. 1988, 66, 792–795, doi:10.1139/o88-090.
- Parlo, R.A.; Coleman, P.S. Continuous pyruvate carbon flux to newly synthesized cholesterol and the suppressed evolution of pyruvate-generated CO2 in tumors: Further evidence for a persistent truncated Krebs cycle in hepatomas. Biochim. Biophys. Acta 1986, 886, 169–176, doi:10.1016/0167-4889(86)90134-5.