Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Endocrinology & Metabolism
Steroid sex hormones are classified as androgens, estrogens, and progestogens. Although all three classes are important in male and female physiology, androgens are associated with "musculisation" effects and are considered primarily male sex hormones. Androgens have diverse functions in muscle physiology, lean body mass, the regulation of adipose tissue, bone density, neurocognitive regulation, and spermatogenesis, male reproductive and sexual function.
- male hypogonadism
1. Introduction
Steroid sex hormones are classified as androgens, estrogens, and progestogens. Although all three classes are important in male and female physiology, androgens are associated with "musculisation" effects and are considered primarily male sex hormones [1]. Androgens have diverse functions in muscle physiology, lean body mass, the regulation of adipose tissue, bone density, neurocognitive regulation, and spermatogenesis, male reproductive and sexual function [2].
When testosterone synthesis is impaired, a condition of hypogonadism arises that affects quality of life and wellbeing [3]. Because of the importance of testosterone in male physiology, hypogonadism further leads to increased fat accumulation, a reduction in lean body mass, and osteoporosis. Hypogonadism may also arise as a consequence of the ageing process, which can be described as the gradual deterioration in biological function over time, reducing quality of life and increasing the risk of degenerative noncommunicable chronic diseases (NCDs) [4]. In fact, ageing has an important relationship as both a risk factor and/or a comorbidity with NCDs, including obesity, metabolic syndrome, type-2 diabetes mellitus (T2DM), and numerous malignancies [2,4].
All these conditions have in common an imbalance in the redox homeostasis in favor of higher levels of oxidants, leading to the development of oxidative stress [5]. Oxidative stress is a leading mechanism that drives the ageing process, with a complex relationship in the pathogenesis of age-related NCDs. Moreover, it may have an important role in age-related hypogonadism associated with the increased risk of NCDs [6,7,8].
2. Steroidogenesis and Male Hypogonadism
In males, testosterone is synthesized primarily in Leydig cells through LH receptor (LHR) binding, with the activation of G-coupled protein and adenylyl cyclase, which increases intracellular cyclic adenosine monophosphate (cAMP). This leads to the activation of the steroidogenic acute regulatory (StAR) protein that mediates the mitochondrial uptake of cholesterol, which is converted to pregnenolone by the cytochrome P450 side chain cleavage enzyme [9,10]. In humans, 17α-hydroxylase (CYP17A1) converts pregnenolone to 17α-hydroxy-pregnenolone and androgen dehydroepiandrosterone (DHEA). DHEA is converted to androstenedione via 3β-hydroxysteroid dehydrogenase (3β-HSD), and then to testosterone via 17β-hydroxysteroid dehydrogenase-3 (17β-HSD-3) [9,10]. The steroidogenesis pathway is summarized in Figure 1. cAMP activation is required for the expression of steroidogenesis enzymes [11]. Androgens exist both in free form and bound to serum proteins. Although approximately 98% of testosterone is bound to albumin or sex-hormone-binding globulin (SHBG), about 2% of circulating testosterone is not bound to serum proteins and is able to penetrate into cells and exert its metabolic effects [12].
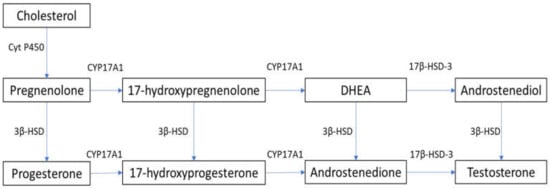
Figure 1. Steroidogenesis pathway. 3β-HSD: 3β-hydroxysteroid dehydrogenase; 17β-HSD-3: 17β-hydroxysteroid dehydrogenase-3; Cyt P450: cytochrome P450; CYP17A1: 17α-hydroxylase; DHEA: dehydroepiandrosterone.
Male hypogonadism is a clinical syndrome caused by a disruption of the hypothalamic–pituitary–gonadal (HPG) axis that affects the testicular synthesis of testosterone [3]. Various terminologies are used to describe this syndrome, including testicular failure, androgen deficiency syndrome, testosterone deficiency syndrome, andropause, androgen deficiency in ageing males (ADAM), and late-onset hypogonadism (LOH) [3]. Hypogonadism is estimated to affect up to 12% of male adults in the general population, and the incidence is expected to increase in the future [3], mainly because of the rise in the population aged 65 years and over.
The classification of hypogonadism is based on testicular or non-testicular causes. Testicular failure is classified as primary (hypergonadotropic) hypogonadism, and the causes include Klinefelter syndrome, Sertoli-cell-only syndrome, cryptorchidism, testicular trauma, mumps orchitis, radiation or chemotherapy treatment, and autoimmune diseases [3]. Pituitary or hypothalamic causes are classified as secondary (hypogonadotropic) hypogonadism, and include Kalman’s syndrome, pituitary adenoma, hyperprolactinaemia, and medications [3]. Although often reported as secondary hypogonadism, the nontesticular causes of "mixed" hypogonadism can be caused by ageing, excessive alcohol consumption, and corticosteroid treatment [3]. Important clinical associations with hypogonadism as risk factors and/or comorbidities include obesity, metabolic syndrome, T2DM, cardiovascular disease, and osteoporosis [3]. Furthermore, lower levels of testosterone in healthy men are a predictor of obesity, metabolic syndrome, and related comorbidities [13]. Hypogonadism is also associated with environmental exposures that induce oxidative stress, which can result in male infertility. This includes exposure to air pollution, pesticides, heavy metals, radiation, and, particularly, endocrine-disrupting chemicals [14].
Hypogonadism presents clinically with sexual dysfunction, prominently including erectile dysfunction, infertility, increased adiposity with decreased muscle mass, reduced bone density, and osteoporosis, fatigue, and depression [3]. Diagnosis is confirmed with a reduced serum total testosterone on two separate occasions [15], while the determination of the serum LH differentiates primary (increased LH: hypergonadotropic) from secondary (reduced LH: hypogonadotropic) hypogonadism [15]. Age-associated hypogonadism may be characterized by normal or low-normal levels of LH [15].
3. Oxidative Stress and Hypogonadism
In living cells, redox (reduction and oxidation) reactions mediate numerous physiological pathways; hence, the intracellular levels of oxidants and antioxidants play an important role in this fine regulation [5,16]. Reactive oxygen species (ROS) are oxygen-based oxidants that are generated during cellular metabolism, predominantly in the mitochondria. They act as physiological mediators in several processes, such as immune regulation, inflammation, apoptosis, and the regulation of genetic expression, among others [5,16]. The ROS family includes both radical and nonradical species (Figure 2).
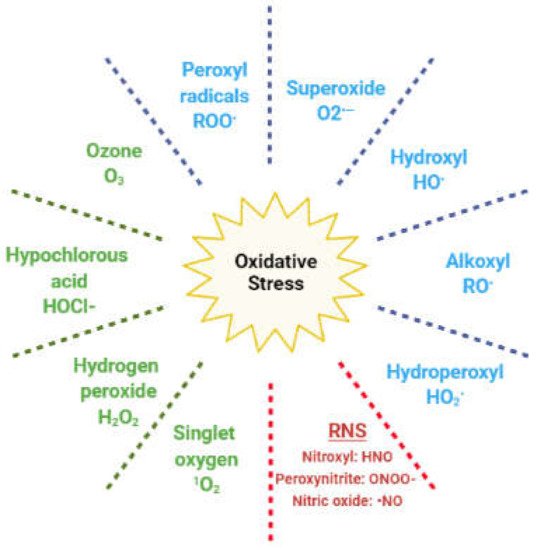
Figure 2. Reactive species responsible for oxidative stress. In blue: radical oxygen species; in green: nonradical oxygen species; in red: reactive nitrogen species (RNS).
The former are molecules with unpaired electrons in the outer orbit; hence, they easily react with any other cellular molecule, including lipids, proteins, and DNA. Nonradical species include hydrogen peroxide (H2O2), which can react with ferrous ions in the Fenton reaction and lead to the generation of hydroxyl radical. Other oxidants originate from nitrogen and are classified as reactive nitrogen species (RNS) [5]. Redox homeostasis is maintained by enzymatic and nonenzymatic antioxidant compounds, which are endogenously generated, or introduced exogenously through the diet (Table 1) [17].
Table 1. Enzymatic and nonenzymatic antioxidants.
| Enzymatic | Nonenzymatic |
|---|---|
| Superoxide dismutase (SOD) | Vitamin C, vitamin E, vitamin B9 |
| Catalase (CAT) | Selenium, Zinc, Mn2+ |
| Glutathione peroxidase (GPx) | Carotenoids, flavonoids, lycopene |
| Glutathione reductase (GR) | Taurine, hypotaurine |
| Glutathione-S-transferase (GST) | Glutathione, inositol, cysteine, coenzyme Q10 |
| Thioredoxin |
When the fine redox equilibrium is shifted in favour of oxidants through increased ROS or reduced antioxidants, a condition of oxidative stress arises. As numerous cellular signaling pathways respond to variations in redox status, oxidative stress can consequently result in the disruption of the cellular signalling and cause cellular damage [18]. Several studies have described the ROS-dependent regulation of molecular pathways, such as extracellular signal-regulated kinases (ERK), c-Jun N-terminal kinases (JNK), phosphatidylinositol 3-Kinase (PI3K)/Akt, p38, p53, protein-kinase C, phospholipase C, nuclear factor kB (NF kB), and JAK/STAT. These can have either a pro- or anti-apoptotic effect (for review on the topic, see [19,20]. In clinical practice, oxidative stress has been widely described as a contributing factor in several pathological conditions, such as cardiovascular, autoimmune, and neurodegenerative disorders, as well as cancer, diabetes, and infertility [21,22,23,24,25].
ROS are physiologically produced during the enzymatic reactions of steroidogenesis. Monooxygenase reactions require electron donation from NADPH through adrenodoxin reductase and adrenodoxin [26,27]. Here, electron leakage results in the generation of superoxide and other ROS [26,28]. Experiments in a mouse Leydig tumor (MA-10) cell line demonstrated that ROS mediate the cAMP-activation of RAS and the phosphorylation of ERK1/2, after the binding of the LH hormone on Leydig cell receptors [29]. The activation of these pathways is reported to positively modulate the proliferation and survival of Leydig cells, as well as steroidogenesis [30,31].
However, a switch of the redox status towards oxidative stress can affect steroidogenesis, resulting in the reduced synthesis of androgens (Figure 3).
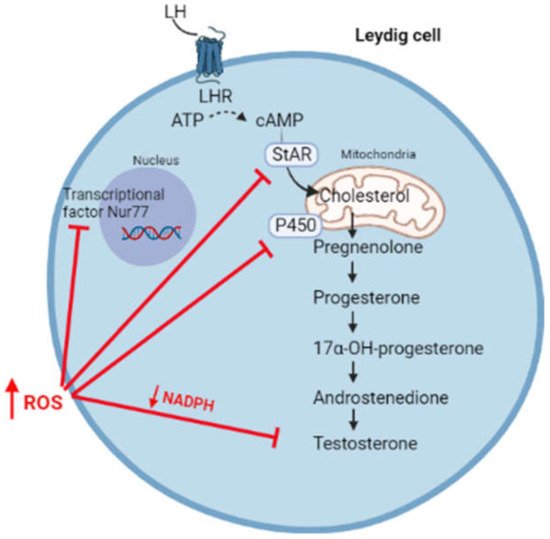
Figure 3. High levels of ROS affect steroidogenesis through the inhibition of transcriptional factor Nur77, the synthesis of StAR, enzymatic P450 activity, and by reducing the levels of NADPH cofactor.
High levels of ROS hyperactivates the JNK signalling, which suppresses the activity of Nur77, a transcriptional factor regulating the expression of several steroidogenic enzymes [32]. Moreover, free radicals can oxidize the heme catalytic group of cytochrome P450, resulting in enzymatic inactivation [33,34]. By using oxidative agents, Chen et al. reported an increased phosphorylation of the MAPK family members (ERK1/2, JNK, and p38) associated with reduced testosterone production in MA-10 cells, highlighting the importance of the redox status in the regulation of steroidogenesis [35]. In this regard, it has been suggested that the activation of the p38 pathway is responsible for reduced testosterone production, possibly through the activation of cyclo-oxygenase2 (COX2) [35,36,37]. In fact, the overexpression of COX-2 has been associated with the reduced expression of StAR and, consequently, with reduced testosterone synthesis [38,39]. Moreover, high levels of ROS are responsible for the depolarization of the mitochondrial membrane, associated with the post-transcriptional inhibition of the StAR protein [40].
ROS scavenging relies on antioxidant systems. The reduced synthesis of testosterone was observed in Nrf2 knock-out mice, where Nrf2 is a transcription factor regulating the expression of antioxidant systems in response to oxidative stress [41]. Experiments conducted in young and aged rats showed that the depletion of glutathione (GSH) was accompanied by reduced testosterone synthesis [42]. In addition, the switch towards an oxidative status with reduced levels of NADPH can indirectly affect steroidogenesis through the inhibition of enzymatic activities. In fact, NADPH is an important cofactor of several endogenous antioxidants, such as GSH and thioredoxin, as well as enzymes involved in steroidogenesis [10,43].
This entry is adapted from the peer-reviewed paper 10.3390/antiox10111834
This entry is offline, you can click here to edit this entry!