Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
The standard treatment of locally advanced esophageal cancer comprises multimodal treatment concepts including preoperative chemoradiotherapy (CRT) followed by radical surgical resection. However, despite intensified treatment approaches, 5-year survival rates are still low. Therefore, new strategies are required to overcome treatment resistance, and to improve patients’ outcome.
- esophageal cancer
- chemoradiotherapy
- treatment resistance
- chemoradiotherapy-sensitization
- Wnt/β-catenin pathway
1. Introduction
Esophageal cancer is a leading cause of cancer-related morbidity and mortality, with 19,260 estimated new cases and 15,530 estimated deaths in the US in 2021, and 5-year overall survival rates of around 20% [1]. In locally advanced stages of the disease, the standard treatment typically involves a multidisciplinary approach. Depending on the underlying histology (squamous-cell carcinoma (SCC) or adenocarcinoma (AC)), and factors such as localization of the tumor and comorbidities of the patient, either preoperative chemoradiotherapy (CRT) or perioperative chemotherapy (CT) are applied, both followed by radical surgical resection, or definitive CRT without surgery [2][3]. However, the response to pre- or perioperative treatment modalities is heterogeneous, and ranges from complete histopathological regression to complete resistance. This has resulted in significant efforts and clinical trials investigating different multimodal treatment strategies [4][5][6]. A prime example is the CROSS trial, which demonstrated that the median overall survival (OS) was significantly higher in the CRT-plus-surgery group compared to the surgery-only group (48.6 months versus 24.0 months, respectively) [7][8]. Very recently, the follow-up data from the CROSS trial were published, with a 10-year OS of 38% for patients in the CRT-plus-surgery group compared with 25% for patients in the surgery-only group, respectively [9]. Nevertheless, many patients do not benefit from the advances in current treatment strategies, but are exposed to the potential side effects of both CT and irradiation. Accordingly, alternative concepts for patient stratification and novel treatment strategies are urgently needed [10][11]. This is particularly important as patients with preoperative treatment concepts, or relevant comorbidities who are unable to undergo surgical resection, highly rely on the efficacy of CRT, as this represents the only therapeutic choice.
2. Esophageal Cancer Cell Lines Show Different Wnt/β-Catenin Pathway Activities
We and others have demonstrated that aberrant Wnt/β-catenin signaling plays an important role in mediating resistance to different treatment modalities, including CRT [12][13][14][15]. To assess the influence of Wnt/β-catenin signaling in esophageal cancer cells, we first determined the expression levels of relevant Wnt-related proteins such as Axin2, active β-catenin (ABC), total β-catenin (TBC), and the Wnt-transcription factor TCF7L2 by Western blot analysis (Figure 1A). Since there are two main histological subtypes of esophageal cancer, five AC and four SCC cell lines were included in this study. Figure 1A shows that the expression levels of Wnt-related proteins differ among the cell lines, with relatively high expression of active and total β-catenin in the AC cell lines OAC-P4C, SK-GT4, and FLO-1, and the SCC cell lines Kyse-180 and Kyse-150. The Wnt-transcription factor TCF7L2 is expressed in all cell lines, but with varying intensities. The variable expression levels of Wnt-related proteins prompted us to investigate a different transcriptional output of the Wnt/β-catenin pathway. Therefore, we applied TOPFlash/FOPFlash reporter assays to assess the Wnt/β-catenin activity represented by the transcriptional TCF/LEF reporter activity, as a standard in the field [16]. Due to low transfection efficacy, we were not able to analyze Kyse-70 and Kyse-270 cells. Basal Wnt/β-catenin activity was detected in two AC cell lines, OE-19 and OE-33 (Figure 1B). OAC-P4C, FLO-1, Kyse-150, and Kyse-180 showed no basal activity (Figure 1B). To test if exogenous induction of Wnt/β-catenin activity triggers a transcription reporter signal, we assessed reporter activity after transfection of an expression plasmid encoding for a mutated, constitutively active version of β-catenin (S33Y), which cannot be degraded by GSK3β phosphorylation [17]. As displayed in Figure 1C, the high basal TCF/LEF reporter activity of OE-19 cells was further increased (800-fold). Except for OAC-P4C, the other cell lines showed inducible reporter activity.
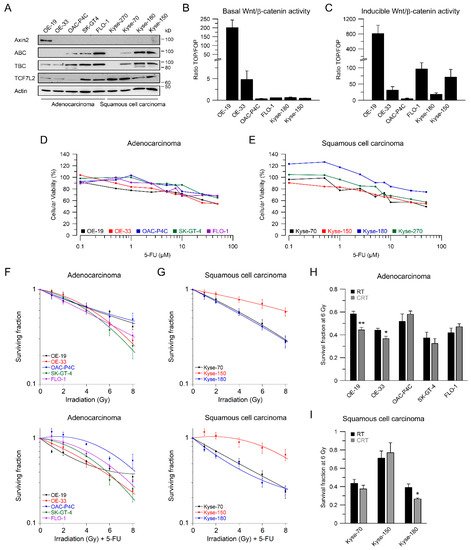
Figure 1. Characterization of Wnt/β-catenin pathway activity, and irradiation sensitivity of esophageal cancer cell lines. (A) Nine esophageal cancer cell lines were analyzed for expression of Wnt-related proteins by immunoblotting; (B,C) Selected cell lines were analyzed for basal Wnt/β-catenin transcriptional activity, (B) or inducible transcriptional activity (C), measured by dual luciferase reporter assays; (D,E) Dose-response analysis for different concentrations of 5-FU 24 h after treatment in adenocarcinoma (D) or squamous cell carcinoma cell lines (E); (F,G) Adenocarcinoma (F) or squamous cell carcinoma cell lines (G) were cultured in colony formation assays (CFA) to determine their survival following irradiation (RT, upper panel) or irradiation in the presence of 5-FU (CRT, lower panel); (H,I) Comparison of RT and CRT survival fractions at 6 Gy. Data are presented as mean ± s.e.m. from at least n = 3 independent biological replicates. * p < 0.05, ** p < 0.01, unpaired two-sample Student’s t-test.
3. RNAi-Mediated Inhibition of Wnt/β-Catenin Signaling Sensitizes to CRT
Beta-catenin is the key intracellular signal transducer of canonical Wnt signaling, which, in the absence of Wnt-ligands, is targeted for degradation by a multi-protein destruction complex consisting mainly of adenomatous polyposis coli (APC), Axin, glycogen synthase kinase 3 beta (GSK3β), and casein kinase 1 alpha (CK1α) [18][19]. Pathway activation occurs through ligand-receptor binding followed by the release of β-catenin from the disintegrated destruction complex. Stabilized β-catenin accumulates in the cytosol, translocates to the nucleus, and subsequently binds to the transcription factors T cell factor (TCF) and lymphoid enhancer–binding factor 1 (LEF1), which in turn up-regulates β-catenin/TCF target genes [19][20][21]. Due to its function as key signal regulator of the canonical Wnt pathway, we silenced β-catenin using RNAi. Depletion of β-catenin resulted in a suppression of both active and total β-catenin protein levels (Figure 2A–F, upper left). Of note, active β-catenin was inconsistently detected in OE-33 cells, which may be due to the lower expression levels. RNAi-mediated silencing of β-catenin had no effect on either cellular viability (Figure 2A–F, upper right), or plating efficiency. As shown in Figure 2A,B, treatment with CRT after RNAi resulted in a significant reduction of clonogenic survival in Wnt/β-catenin active OE-19 cells (RERsiRNA#1 = 1.37, RERsiRNA#2 = 1.24 at 4 Gy), and in OE-33 cells (RERsiRNA#1 = 1.32, RERsiRNA#2 = 1.37 at 4 Gy). In contrast, there was no effect in OAC-P4C, FLO-1, Kyse-150, and Kyse-180 cells (Figure 2C–F). These results suggest that the effect of CRT sensitization after pathway inhibition is restricted to AC cells with basal transcriptional Wnt/β-catenin activity.
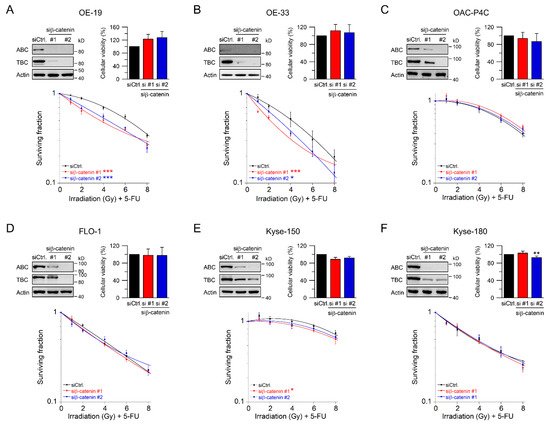
Figure 2. Treatment sensitization following β-catenin depletion depends on the Wnt/β-catenin-pathway activity. (A–F) Signaling-active cell lines OE-19 (A) and OE-33 (B), and signaling-inactive cell lines OAC-P4C (C), FLO-1 (D), Kyse-150 (E), and Kyse-180 (F) were transfected with control siRNA (siCtrl.) or siRNA targeting β-catenin (siβ-catenin #1, #2), and subjected to immunoblot analyses (upper left), and cellular viability assays (upper right). Following siRNA-mediated silencing of β-catenin, cells were monitored for CFA survival after irradiation in the presence of 5-FU (CRT) (lower graph). Data presented as mean ± s.e.m. from at least n = 3 independent biological replicates. * p < 0.05, ** p < 0.01, *** p < 0.001, unpaired two-sample Student’s t-test or two-way analysis of variance (ANOVA).
4. Fractionated Irradiation in Wnt/β-Catenin Signaling-Independent Cell Lines
In the clinical setting, irradiation is delivered in fractionated doses [22]. Therefore, we mimicked this strategy and tested Wnt/β-catenin inhibition in the context of a fractionated CRT setting. Because 1.8–2 Gy represents the typical individual dose of conventional fractionation delivery RT [23]. Prior irradiation, cells were transfected with siRNA. We only implemented this protocol for those four cell lines for which we did not observe a re-sensitization effect, i.e., OAC-P4C, FLO-1, Kyse-150, and Kyse-180 (Figure 2C–F). Successful silencing following RNAi against β-catenin was assessed after each fraction time point, and analyzed by Western blotting (Figure 3A–D, left). As expected, the CRT sensitivity of OAC-P4C, with no basal and no inducible signaling reporter activity (Figure 1B,C), remained unchanged (Figure 3A). Likewise, FLO-1 and Kyse-180, both of which harbor inducible Wnt/β-catenin activity (Figure 1C), revealed no changes in the CRT survival (Figure 3B,C), respectively. As displayed in Figure 3D, the highly resistant SCC cell line Kyse-150 revealed impaired clonogenic survival following depletion of β-catenin in a fractionated treatment regimen (RERsiRNA#1 = 1.6, RERsiRNA#2 = 1.41 at 6 Gy). However, the surviving fraction of Kyse-150 cells remains high with a fractionated irradiation protocol after β-catenin inhibition, which limits the value of targeting the Wnt/β-catenin pathway in this model. At this point, it is important to note that Kyse-150 originates from a patient who was treated with RT [24]. Therefore, this cell line is highly refractory to CRT.
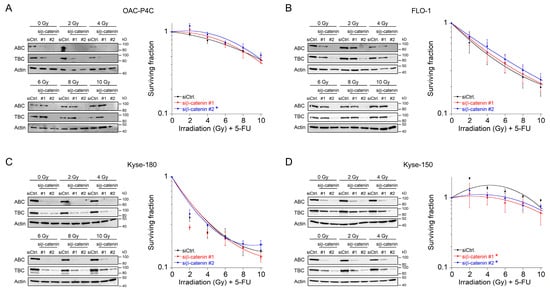
Figure 3. Survival analyses of basal Wnt/β-catenin signaling inactive cells in a fractionated irradiation experimental design. (A–D) Cell lines OAC-P4C (A), FLO-1 (B), Kyse-180 (C), or Kyse-150 (D) were transfected with a control siRNA (siCtrl.), or with siRNA targeting β-catenin (siβ-catenin #1, #2), and subjected to immunoblot analyses (left), or monitored for CFA survival after CRT as fractionated irradiation in doses of 2 Gy every twelve hours (right). Data presented as mean ± s.e.m. from at least n = 3 independent biological replicates. * p < 0.05, two-way analysis of variance (ANOVA).
5. Using Small-Molecule Inhibitors as a Therapeutic Strategy to Sensitize Esophageal Cancer Cells
To evaluate whether targeting Wnt/β-catenin signaling at a pharmacological level represents a potential clinical strategy for Wnt/β-catenin active tumors, we tested XAV-939 [25]. This compound is a small-molecule inhibitor of tankyrase 1 and tankyrase 2, which stabilize Axin as part of the β-catenin destruction complex [19][25]. Western blot analyses demonstrated induction of Axin2 expression and, at the same time, reduction of active and total β-catenin (Figure 4A–E, upper left). In OE-19, OAC-P4C, FLO-1, and Kyse-180 cells, cellular viability was significantly reduced following XAV-939 treatment (Figure 4A–E, upper right). However, this reduction of cellular viability had no effect on colony formation and did not limit irradiation experiments. The most striking effect of XAV-939 treatment on survival after CRT was observed in OE-19 cells, with an RER2.5 µM XAV-939 = 1.61 and an RER5 µM XAV-939 = 1.56 at 4 Gy (Figure 4A). This effect was even higher compared to the effect observed after CRT following RNAi-mediated depletion of β-catenin (Figure 2A). In Wnt/β-catenin active OE-33 cells, treatment with XAV-939 resulted in decreased CRT survival rates with an RER5 µM XAV-939 = 1.39 and an RER10 µM XAV-939 = 1.23 at 4 Gy (Figure 4B). In none of the other cell lines (Figure 4C–E), CFA survival was affected after treatment with XAV-939.
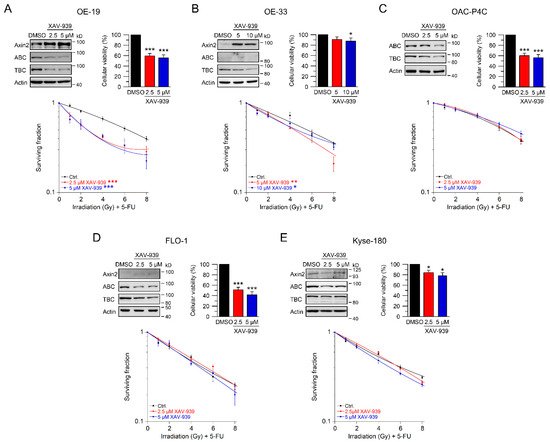
Figure 4. Inhibition of Wnt/β-catenin signaling by the tankyrase inhibitor XAV-939. (A–E) Cell lines were left untreated (DMSO), or treated with two different concentrations of the tankyrase inhibitor XAV-939. Cell lines ((A)—OE-19, (B)—OE-33, (C)—OAC-P4C, (D)—FLO-1, (E)—Kyse-180) were first subjected to Western blot analysis (upper left), or cellular viability assays (upper right), or were monitored for CFA survival after CRT (lower graphs). Data presented as mean ± s.e.m. from at least n = 3 independent biological replicates. * p < 0.05, ** p < 0.01, *** p < 0.001, two-way analysis of variance (ANOVA).
To further confirm these results, we used JW55, another tankyrase inhibitor. In contrast to XAV-939, this small-molecule inhibitor can be administered orally [26]. Again, we first established reasonable doses and time points for JW55. Upon treatment with two doses of JW55, OE-19, OE-33, FLO-1, Kyse-150, and Kyse-180 showed increased Axin2 levels and reduced active and total β-catenin protein expression (Figure 5A–E, upper left). In general, cellular viability of JW55 treated cells was not affected when compared to the DMSO control (Figure 5B–E, upper right). Only OE-19 cells revealed an impaired cellular viability (Figure 5A upper right), which did not interfere with the ability to form colonies. In clonogenic survival assays, treatment of OE-19 and OE-33 cells with JW55 rendered both cell lines more sensitive to CRT, as revealed by their decreased CFA survival rates with an RER5 µM JW55 = 1.29 and an RER10 µM JW55 = 1.54 at 4 Gy for OE-19 (Figure 5A) and an RER10 µM JW55 = 1.39 at 4 Gy for OE-33 (Figure 5B). Note that none of the JW55 concentrations affected CFA survival of Wnt/β-catenin inactive cell lines FLO-1, Kyse-150, and Kyse-180 (Figure 5C–E). In summary, only cell lines with relevant basal transcriptional Wnt/β-catenin activity could be (re-) sensitized to CRT upon tankyrase/β-catenin inhibition, either by XAV-939 or JW55.
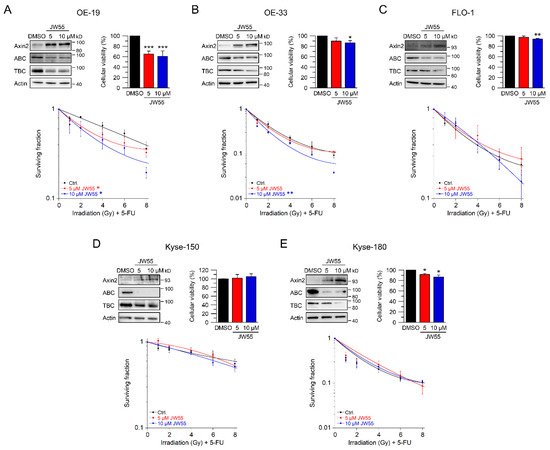
Figure 5. Inhibition of Wnt/β-catenin signaling by the tankyrase inhibitor JW55. (A–E) Cell lines were left untreated (DMSO), or treated with two different concentrations of the tankyrase inhibitor JW55. Cell lines ((A)—OE-19, (B)—OE-33, (C)—FLO-1, (D)—Kyse-150, (E)—Kyse-180) were first subjected to Western blot analysis (upper left), or cellular viability assays (upper right), or were monitored for CFA survival after CRT (lower graphs). Data presented as mean ± s.e.m. from at least n = 3 independent biological replicates. * p < 0.05, ** p < 0.01, *** p < 0.001, two-way analysis of variance (ANOVA).
This entry is adapted from the peer-reviewed paper 10.3390/ijms221910301
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33.
- Kelly, R.J. Emerging Multimodality Approaches to Treat Localized Esophageal Cancer. J. Natl. Compr. Cancer Netw. 2019, 17, 1009–1014.
- Lagergren, J.; Smyth, E.; Cunningham, D.; Lagergren, P. Oesophageal cancer. Lancet 2017, 390, 2383–2396.
- Al-Batran, S.E.; Homann, N.; Pauligk, C.; Goetze, T.O.; Meiler, J.; Kasper, S.; Kopp, H.G.; Mayer, F.; Haag, G.M.; Luley, K.; et al. Perioperative chemotherapy with fluorouracil plus leucovorin, oxaliplatin, and docetaxel versus fluorouracil or capecitabine plus cisplatin and epirubicin for locally advanced, resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4): A randomised, phase 2/3 trial. Lancet 2019, 393, 1948–1957.
- Allum, W.H.; Stenning, S.P.; Bancewicz, J.; Clark, P.I.; Langley, R.E. Long-term results of a randomized trial of surgery with or without preoperative chemotherapy in esophageal cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 5062–5067.
- Cunningham, D.; Allum, W.H.; Stenning, S.P.; Thompson, J.N.; Van de Velde, C.J.; Nicolson, M.; Scarffe, J.H.; Lofts, F.J.; Falk, S.J.; Iveson, T.J.; et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N. Engl. J. Med. 2006, 355, 11–20.
- Shapiro, J.; van Lanschot, J.J.B.; Hulshof, M.; van Hagen, P.; van Berge Henegouwen, M.I.; Wijnhoven, B.P.L.; van Laarhoven, H.W.M.; Nieuwenhuijzen, G.A.P.; Hospers, G.A.P.; Bonenkamp, J.J.; et al. Neoadjuvant chemoradiotherapy plus surgery versus surgery alone for oesophageal or junctional cancer (CROSS): Long-term results of a randomised controlled trial. Lancet Oncol. 2015, 16, 1090–1098.
- van Hagen, P.; Hulshof, M.C.; van Lanschot, J.J.; Steyerberg, E.W.; van Berge Henegouwen, M.I.; Wijnhoven, B.P.; Richel, D.J.; Nieuwenhuijzen, G.A.; Hospers, G.A.; Bonenkamp, J.J.; et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N. Engl. J. Med. 2012, 366, 2074–2084.
- Eyck, B.M.; van Lanschot, J.J.B.; Hulshof, M.; van der Wilk, B.J.; Shapiro, J.; van Hagen, P.; van Berge Henegouwen, M.I.; Wijnhoven, B.P.L.; van Laarhoven, H.W.M.; Nieuwenhuijzen, G.A.P.; et al. Ten-Year Outcome of Neoadjuvant Chemoradiotherapy Plus Surgery for Esophageal Cancer: The Randomized Controlled CROSS Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 1995–2004.
- Davies, A.R.; Myoteri, D.; Zylstra, J.; Baker, C.R.; Wulaningsih, W.; Van Hemelrijck, M.; Maisey, N.; Allum, W.H.; Smyth, E.; Gossage, J.A.; et al. Lymph node regression and survival following neoadjuvant chemotherapy in oesophageal adenocarcinoma. Br. J. Surg. 2018, 105, 1639–1649.
- Noble, F.; Lloyd, M.A.; Turkington, R.; Griffiths, E.; O’Donovan, M.; O’Neill, J.R.; Mercer, S.; Parsons, S.L.; Fitzgerald, R.C.; Underwood, T.J. Multicentre cohort study to define and validate pathological assessment of response to neoadjuvant therapy in oesophagogastric adenocarcinoma. Br. J. Surg. 2017, 104, 1816–1828.
- Emons, G.; Spitzner, M.; Reineke, S.; Moller, J.; Auslander, N.; Kramer, F.; Hu, Y.; Beissbarth, T.; Wolff, H.A.; Rave-Frank, M.; et al. Chemoradiotherapy Resistance in Colorectal Cancer Cells is Mediated by Wnt/beta-catenin Signaling. Mol. Cancer Res. 2017, 15, 1481–1490.
- Kendziorra, E.; Ahlborn, K.; Spitzner, M.; Rave-Fränk, M.; Emons, G.; Gaedcke, J.; Kramer, F.; Wolff, H.A.; Becker, H.; Beissbarth, T.; et al. Silencing of the Wnt transcription factor TCF4 sensitizes colorectal cancer cells to (chemo-) radiotherapy. Carcinogenesis 2011, 32, 1824–1831.
- Kumar, V.; Vashishta, M.; Kong, L.; Wu, X.; Lu, J.J.; Guha, C.; Dwarakanath, B.S. The Role of Notch, Hedgehog, and Wnt Signaling Pathways in the Resistance of Tumors to Anticancer Therapies. Front. Cell Dev. Biol. 2021, 9, 650772.
- Zhao, Y.; Tao, L.; Yi, J.; Song, H.; Chen, L. The Role of Canonical Wnt Signaling in Regulating Radioresistance. Cell Physiol. Biochem. Int. J. Exp. Cell Physiol. Biochem. Pharmacol. 2018, 48, 419–432.
- Korinek, V.; Barker, N.; Morin, P.J.; van Wichen, D.; de Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/-colon carcinoma. Science 1997, 275, 1784–1787.
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997, 275, 1787–1790.
- Chatterjee, A.; Paul, S.; Bisht, B.; Bhattacharya, S.; Sivasubramaniam, S.; Paul, M.K. Advances in targeting the WNT/β-catenin signaling pathway in cancer. Drug Discov. Today 2021.
- Jackstadt, R.; Hodder, M.C.; Sansom, O.J. WNT and beta-Catenin in Cancer: Genes and Therapy. Annu. Rev. Cancer Biol. 2020, 4, 177–196.
- Clevers, H.; Loh, K.M.; Nusse, R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014, 346, 6205–1248012.
- Nusse, R. Wnt signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011163.
- Paireder, M.; Jomrich, G.; Kristo, I.; Asari, R.; Rieder, E.; Beer, A.; Ilhan-Mutlu, A.; Preusser, M.; Schmid, R.; Schoppmann, S.F. Modification of preoperative radiochemotherapy for esophageal cancer (CROSS protocol) is safe and efficient with no impact on surgical morbidity. Strahlenther. Onkol. 2020, 196, 779–786.
- Subiel, A.; Ashmore, R.; Schettino, G. Standards and Methodologies for Characterizing Radiobiological Impact of High-Z Nanoparticles. Theranostics 2016, 6, 1651–1671.
- Shimada, Y.; Imamura, M.; Wagata, T.; Yamaguchi, N.; Tobe, T. Characterization of 21 newly established esophageal cancer cell lines. Cancer 1992, 69, 277–284.
- Huang, S.M.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620.
- Waaler, J.; Machon, O.; Tumova, L.; Dinh, H.; Korinek, V.; Wilson, S.R.; Paulsen, J.E.; Pedersen, N.M.; Eide, T.J.; Machonova, O.; et al. A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res. 2012, 72, 2822–2832.
This entry is offline, you can click here to edit this entry!