Breast cancer is the most frequently diagnosed cancer and the primary cause of cancer death in women worldwide. Although early diagnosis and cancer growth inhibition has significantly improved breast cancer survival rate over the years, there is a current need to develop more effective systemic treatments to prevent metastasis. One of the most commonly altered pathways driving breast cancer cell growth, survival, and motility is the PI3K/AKT/mTOR signaling cascade.
Phosphoinositide 3-kinase (PI3K) is a group of lipid kinases that phosphorylate the 3′-OH group of phosphatidylinositol (PI) at plasma and intracellular membranes.
- PI3K
- inhibitor
- AKT
- mTOR
- breast cancer
1. Introduction
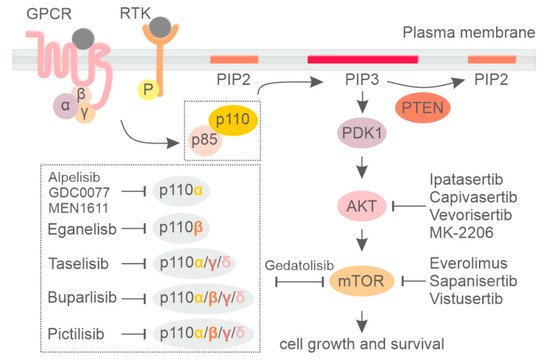
2. PI3K Isoform-Specific Inhibitors
-
Alpelisib
-
Taselisib
3. PI3K Pathway Inhibition in HER2+ and Triple-Negative Breast Cancer Subtypes
-
HER2-Positive Breast Cancer
-
Triple-Negative Breast Cancer (TNBC)
4. Currently Available Inhibitors Acting on AKT and mTOR in Breast Cancer
-
AKT Inhibitors
-
mTOR Inhibitors
-
Dual PI3K/mTOR Inhibitors
5. Rationale for Targeting Class II PI3K in Breast Cancer
This entry is adapted from the peer-reviewed paper 10.3390/cancers13143517
References
- Bilanges, B.; Posor, Y.; Vanhaesebroeck, B. PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell Biol. 2019, 20, 515–534.
- Gulluni, F.; De Santis, M.C.; Margaria, J.P.; Martini, M.; Hirsch, E. Class II PI3K Functions in Cell Biology and Disease. Trends Cell Biol. 2019, 29, 339–359.
- Hirsch, E.; Gulluni, F.; Martini, M. Phosphoinositides in cell proliferation and metabolism. Adv. Biol. Regul. 2020, 75, 100693.
- Maehama, T.; Dixon, J.E. The Tumor Suppressor, PTEN/MMAC1, Dephosphorylates the Lipid Second Messenger, Phosphatidylinositol 3,4,5-Trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378.
- Geering, B.; Cutillas, P.R.; Nock, G.; Gharbi, S.I.; Vanhaesebroeck, B. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc. Natl. Acad. Sci. USA 2007, 104, 7809–7814.
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341.
- Kok, K.; Geering, B.; Vanhaesebroeck, B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem. Sci. 2009, 34, 115–127.
- Kok, K.; Nock, G.E.; Verrall, E.A.G.; Mitchell, M.P.; Hommes, D.W.; Peppelenbosch, M.; Vanhaesebroeck, B. Regulation of p110δ PI 3-Kinase Gene Expression. PLoS ONE 2009, 4, e5145.
- Samuels, Y.; Velculescu, V. Oncogenic Mutations of PIK3CA in Human Cancers. Cell Cycle 2004, 3, 1221–1224.
- Lindhurst, M.J.; Parker, V.E.R.; Payne, F.; Sapp, J.; Rudge, S.; Harris, J.; Witkowski, A.M.; Zhang, Q.; Groeneveld, M.P.; Scott, C.E.; et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat. Genet. 2012, 44, 928–933.
- Angulo, I.; Vadas, O.; Garçon, F.; Banham-Hall, E.; Plagnol, V.; Leahy, T.R.; Baxendale, H.; Coulter, T.; Curtis, J.; Wu, C.; et al. Phosphoinositide 3-Kinase Gene Mutation Predisposes to Respiratory Infection and Airway Damage. Science 2013, 342, 866–871.
- Lucas, C.; Kuehn, H.S.; Zhao, F.; Niemela, J.; Deenick, E.K.; Palendira, U.; Avery, D.T.; Moens, L.; Cannons, J.L.; Biancalana, M.; et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat. Immunol. 2014, 15, 88–97.
- Lucas, C.; Zhang, Y.; Venida, A.; Wang, Y.; Hughes, J.; McElwee, J.; Butrick, M.; Matthews, H.; Price, S.; Biancalana, M.; et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J. Exp. Med. 2014, 211, 2537–2547.
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635.
- Vanhaesebroeck, B.; Perry, M.W.D.; Brown, J.R.; André, F.; Okkenhaug, K. PI3K inhibitors are finally coming of age. Nat. Rev. Drug Discov. 2021, 1–29.
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88.
- Dienstmann, R.; Rodon, J.; Serra, V.; Tabernero, J. Picking the Point of Inhibition: A Comparative Review of PI3K/AKT/mTOR Pathway Inhibitors. Mol. Cancer Ther. 2014, 13, 1021–1031.
- Fritsch, C.; Huang, A.; Chatenay-Rivauday, C.; Schnell, C.; Reddy, A.; Liu, M.; Kauffmann, A.; Guthy, D.; Erdmann, D.; De Pover, A.; et al. Characterization of the Novel and Specific PI3Kα Inhibitor NVP-BYL719 and Development of the Patient Stratification Strategy for Clinical Trials. Mol. Cancer Ther. 2014, 13, 1117–1129.
- Fritsch, C.; Pfister, E.; Ebel, N.; Guthy, D.; Schnell, C.; Hofmann, F. Abstract 3934: Determination of the PI3Kα selective inhibitor alpelisib mechanism of action and efficacy in ER+/PIK3CA mutant breast cancer preclinical models. Exp. Mol. Ther. 2018, 78, 3934.
- Miller, T.W.; Hennessy, B.T.; González-Angulo, A.M.; Fox, E.M.; Mills, G.B.; Chen, H.; Higham, C.; García-Echeverría, C.; Shyr, Y.; Arteaga, C.L. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor–positive human breast cancer. J. Clin. Investig. 2010, 120, 2406–2413.
- Crowder, R.J.; Phommaly, C.; Tao, Y.; Hoog, J.; Luo, J.; Perou, C.; Parker, J.S.; Miller, M.A.; Huntsman, D.G.; Lin, L.; et al. PIK3CA and PIK3CB Inhibition Produce Synthetic Lethality when Combined with Estrogen Deprivation in Estrogen Receptor–Positive Breast Cancer. Cancer Res. 2009, 69, 3955–3962.
- Juric, D.; Janku, F.; Rodón, J.; Burris, H.A.; Mayer, I.A.; Schuler, M.; Seggewiss-Bernhardt, R.; Gil-Martin, M.; Middleton, M.R.; Baselga, J.; et al. Alpelisib Plus Fulvestrant in PIK3CA-Altered and PIK3CA-Wild-Type Estrogen Receptor–Positive Advanced Breast Cancer. JAMA Oncol. 2019, 5, e184475.
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940.
- Moehler, M.; Shitara, K.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Bragagnoli, A.C.; Liu, T.; et al. LBA6_PR Nivolumab (nivo) plus chemotherapy (chemo) versus chemo as first-line (1L) treatment for advanced gastric cancer/gastroesophageal junction cancer (GC/GEJC)/esophageal adenocarcinoma (EAC): First results of the CheckMate 649 study. Ann. Oncol. 2020, 31, S1191.
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936.
- Im, S.-A.; Lu, Y.-S.; Bardia, A.; Harbeck, N.; Colleoni, M.; Franke, F.; Chow, L.; Sohn, J.; Lee, K.-S.; Campos-Gomez, S.; et al. Overall Survival with Ribociclib plus Endocrine Therapy in Breast Cancer. N. Engl. J. Med. 2019, 381, 307–316.
- Rugo, H.S.; Lerebours, F.; Ciruelos, E.; Drullinsky, P.; Borrego, M.R.; Neven, P.; Park, Y.H.; Prat, A.; Bachelot, T.; Juric, D.; et al. Alpelisib (ALP) + fulvestrant (FUL) in patients (pts) with PIK3CA-mutated (mut) hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2–) advanced breast cancer (ABC) previously treated with cyclin-dependent kinase 4/6 inhibitor (CDKi) + aromatase inhibitor (AI): BYLieve study results. J. Clin. Oncol. 2020, 38, 1006.
- Baselga, J.; Cortes, J.; Im, S.-A.; Clark, E.; Ross, G.; Kiermaier, A.; Swain, S. Biomarker Analyses in CLEOPATRA: A Phase III, Placebo-Controlled Study of Pertuzumab in Human Epidermal Growth Factor Receptor 2–Positive, First-Line Metastatic Breast Cancer. J. Clin. Oncol. 2014, 32, 3753–3761.
- Olivero, A.G.; Heffron, T.P.; Baumgardner, M.; Belvin, M.; Ross, L.B.; Blaquiere, N.; Bradley, E.; Castanedo, G.; Derynck, M.; Do, S.; et al. Abstract DDT02-01: Discovery of GDC-0032: A beta-sparing PI3K inhibitor active against PIK3CA mutant tumors. Cancer Chem. 2013, 73.
- Juric, D.; Krop, I.; Ramanathan, R.K.; Wilson, T.R.; Ware, J.A.; Bohorquez, S.S.; Savage, H.M.; Sampath, D.; Salphati, L.; Lin, R.S.; et al. Phase I Dose-Escalation Study of Taselisib, an Oral PI3K Inhibitor, in Patients with Advanced Solid Tumors. Cancer Discov. 2017, 7, 704–715.
- Baselga, J.; Dent, S.F.; Cortés, J.; Im, Y.-H.; Diéras, V.; Harbeck, N.; Krop, I.E.; Verma, S.; Wilson, T.R.; Jin, H.; et al. Phase III study of taselisib (GDC-0032) + fulvestrant (FULV) v FULV in patients (pts) with estrogen receptor (ER)-positive, PIK3CA-mutant (MUT), locally advanced or metastatic breast cancer (MBC): Primary analysis from SANDPIPER. J. Clin. Oncol. 2018, 36, LBA1006.
- Ndubaku, C.O.; Heffron, T.P.; Staben, S.T.; Baumgardner, M.; Blaquiere, N.; Bradley, E.; Bull, R.; Do, S.; Dotson, J.; Dudley, D.; et al. Discovery of 2--2-methylpropanamide (GDC-0032): A β-Sparing Phosphoinositide 3-Kinase Inhibitor with High Unbound Exposure and Robust in Vivo Antitumor Activity. J. Med. Chem. 2013, 56, 4597–4610.
- Hanker, A.; Pfefferle, A.D.; Balko, J.M.; Kuba, M.G.; Young, C.D.; Sánchez, V.; Sutton, C.R.; Cheng, H.; Perou, C.; Zhao, J.J.; et al. Mutant PIK3CA accelerates HER2-driven transgenic mammary tumors and induces resistance to combinations of anti-HER2 therapies. Proc. Natl. Acad. Sci. USA 2013, 110, 14372–14377.
- Loibl, S.; Majewski, I.; Guarneri, V.; Nekljudova, V.; Holmes, E.; Bria, E.; Denkert, C.; Schem, C.; Sotiriou, C.; Loi, S.; et al. Corrections to “PIK3CA mutations are associated with reduced pathological complete response rates in primary HER2-positive breast cancer: Pooled analysis of 967 patients from five prospective trials investigating lapatinib and trastuzumab”. Ann. Oncol. 2019, 30, 1180.
- Guerin, M.; Rezai, K.; Isambert, N.; Campone, M.; Autret, A.; Pakradouni, J.; Provansal, M.; Camerlo, J.; Sabatier, R.; Bertucci, F.; et al. PIKHER2: A phase IB study evaluating buparlisib in combination with lapatinib in trastuzumab-resistant HER2-positive advanced breast cancer. Eur. J. Cancer 2017, 86, 28–36.
- Pistilli, B.; Pluard, T.; Urruticoechea, A.; Farci, D.; Kong, A.; Bachelot, T.; Chan, S.; Han, H.S.; Jerusalem, G.; Urban, P.; et al. Phase II study of buparlisib (BKM120) and trastuzumab in patients with HER2+ locally advanced or metastatic breast cancer resistant to trastuzumab-based therapy. Breast Cancer Res. Treat. 2018, 168, 357–364.
- Loibl, S.; de la Pena, L.; Nekljudova, V.; Zardavas, D.; Michiels, S.; Denkert, C.; Rezai, M.; Bermejo, B.; Untch, M.; Lee, S.C.; et al. Neoadjuvant buparlisib plus trastuzumab and paclitaxel for women with HER2+ primary breast cancer: A randomised, double-blind, placebo-controlled phase II trial (NeoPHOEBE). Eur. J. Cancer 2017, 85, 133–145.
- Jain, S.; Shah, A.N.; Santa-Maria, C.A.; Siziopikou, K.; Rademaker, A.; Helenowski, I.; Cristofanilli, M.; Gradishar, W.J. Phase I study of alpelisib (BYL-719) and trastuzumab emtansine (T-DM1) in HER2-positive metastatic breast cancer (MBC) after trastuzumab and taxane therapy. Breast Cancer Res. Treat. 2018, 171, 371–381.
- Barok, M.; Tanner, M.; Köninki, K.; Isola, J. Trastuzumab-DM1 causes tumour growth inhibition by mitotic catastrophe in trastuzumab-resistant breast cancer cells in vivo. Breast Cancer Res. 2011, 13, R46.
- Zhang, M.; Jang, H.; Nussinov, R. PI3K inhibitors: Review and new strategies. Chem. Sci. 2020, 11, 5855–5865.
- Hong, R.; Edgar, K.; Song, K.; Steven, S.; Young, A.; Hamilton, P.; Arrazate, A.; De La Cruz, C.; Chan, C.; Pang, J.; et al. Abstract PD4-14: GDC-0077 is a selective PI3Kalpha inhibitor that demonstrates robust efficacy in PIK3CA mutant breast cancer models as a single agent and in combination with standard of care therapies. Poster Discuss. Abstr. 2018, 78.
- Turner, N.; Dent, R.; O’Shaughnessy, J.; Kim, S.-B.; Isakoff, S.; Barrios, C.; Saji, S.; Bondarenko, I.; Nowecki, Z.; Lian, Q.; et al. 283MO Ipatasertib (IPAT) + paclitaxel (PAC) for PIK3CA/AKT1/PTEN-altered hormone receptor-positive (HR+) HER2-negative advanced breast cancer (aBC): Primary results from Cohort B of the IPATunity130 randomised phase III trial. Ann. Oncol. 2020, 31, S354–S355.
- Merlino, G.; Fiascarelli, A.; Bigioni, M.; Bressan, A.; Irrissuto, C.; Pellacani, A.; Scaltriti, M.; Binaschi, M. Abstract 2160: MEN1611, a novel α-selective PI3K inhibitor in solid tumors. Tumor Biol. 2018, 78, 2160.
- Janku, F.; Huang, H.; Treskova, I.; Pivovarcikova, K.; Call, S.; Meric-Bernstam, F.; Pesta, M.; Polivka, J. Ultra-sensitive detection of circulating tumor DNA identifies patients in high risk of recurrence in early stages melanoma. Ann. Oncol. 2019, 30, v767.
- Hansen, A.R.; Shapiro, G.; Do, K.T.; Kumar, R.; Martin-Liberal, J.; Higano, C.S.; Wisinski, K.B.; Dean, E.J.; Heath, E.I.; Rathkopf, D.E.; et al. A first in human phase I study of AZD8186, a potent and selective inhibitor of PI3K in patients with advanced solid tumours as monotherapy and in combination with the dual mTORC1/2 inhibitor vistusertib (AZD2014) or abiraterone acetate. J. Clin. Oncol. 2017, 35, 2570.
- Owusu-Brackett, N.; Zhao, M.; Akcakanat, A.; Evans, K.W.; Yuca, E.; Dumbrava, E.I.; Janku, F.; Meric-Bernstam, F. Targeting PI3Kβ alone and in combination with chemotherapy or immunotherapy in tumors with PTEN loss. Oncotarget 2020, 11, 969–981.
- Zhang, Z.; Richmond, A. The Role of PI3K Inhibition in the Treatment of Breast Cancer, Alone or Combined With Immune Checkpoint Inhibitors. Front. Mol. Biosci. 2021, 8.
- Szekely, B.; Bossuyt, V.; Li, X.; Wali, V.; Patwardhan, G.; Frederick, C.; Silber, A.; Park, T.; Harigopal, M.; Pelekanou, V.; et al. Immunological differences between primary and metastatic breast cancer. Ann. Oncol. 2018, 29, 2232–2239.
- Hamilton, E.; Lee, A.; Swart, R.; Newton, G.; O’Connell, B.; Roberts, J.; Zhang, H.; Soliman, H. Abstract PS11-32: Mario-3 phase II study safety run-in evaluating a novel triplet combination of eganelisib (formerly IPI-549), atezolizumab (atezo), and nab-paclitaxel (nab-pac) as first-line (1L) therapy for locally advanced or metastatic triple-negative breast cancer (TNBC). Poster Sess. Abstr. 2021, 81, PS11-32.
- Nitulescu, G.M.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Grădinaru, D.; Tsatsakis, A.; Tsoukalas, D.; et al. The Akt pathway in oncology therapy and beyond (Review). Int. J. Oncol. 2018, 53, 2319–2331.
- Bellacosa, A.; Kumar, C.C.; Di Cristofano, A.; Testa, J.R. Activation of AKT Kinases in Cancer: Implications for Therapeutic Targeting. Adv. Cancer Res. 2005, 94, 29–86.
- Banerji, U.; Dean, E.J.; Pérez-Fidalgo, J.A.; Batist, G.; Bedard, P.L.; You, B.; Westin, S.N.; Kabos, P.; Garrett, M.D.; Tall, M.; et al. A Phase I Open-Label Study to Identify a Dosing Regimen of the Pan-AKT Inhibitor AZD5363 for Evaluation in Solid Tumors and inPIK3CA-Mutated Breast and Gynecologic Cancers. Clin. Cancer Res. 2017, 24, 2050–2059.
- De Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arija, J.A.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin. Cancer Res. 2019, 25, 928–936.
- Mundi, P.; Sachdev, J.; McCourt, C.; Kalinsky, K. AKT in cancer: New molecular insights and advances in drug development. Br. J. Clin. Pharmacol. 2016, 82, 943–956.
- Landel, I.; Quambusch, L.; Depta, L.; Rauh, D. Spotlight on AKT: Current Therapeutic Challenges. ACS Med. Chem. Lett. 2020, 11, 225–227.
- Xing, Y.; Lin, N.U.; Maurer, M.A.; Chen, H.; Mahvash, A.; Sahin, A.; Akcakanat, A.; Li, Y.; Abramson, V.; Litton, J.; et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019, 21, 1–12.
- Wright, S.; Vasilevski, N.; Serra, V.; Rodon, J.; Eichhorn, P. Mechanisms of Resistance to PI3K Inhibitors in Cancer: Adaptive Responses, Drug Tolerance and Cellular Plasticity. Cancers 2021, 13, 1538.
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291.
- Weisner, J.; Landel, I.; Reintjes, C.; Uhlenbrock, N.; Trajkovic-Arsic, M.; Dienstbier, N.; Hardick, J.; Ladigan, S.; Lindemann, M.; Smith, S.; et al. Preclinical Efficacy of Covalent-Allosteric AKT Inhibitor Borussertib in Combination with Trametinib in KRAS-mutant Pancreatic and Colorectal Cancer. Cancer Res. 2019, 79, 2367–2378.
- Dey, N.; De, P.; Leyland-Jones, B. PI3K-AKT-mTOR inhibitors in breast cancers: From tumor cell signaling to clinical trials. Pharmacol. Ther. 2017, 175, 91–106.
- Gnant, M.; Baselga, J.; Rugo, H.S.; Noguchi, S.; Burris, H.A.; Piccart, M.; Hortobagyi, G.N.; Eakle, J.; Mukai, H.; Iwata, H.; et al. Effect of Everolimus on Bone Marker Levels and Progressive Disease in Bone in BOLERO-2. J. Natl. Cancer Inst. 2013, 105, 654–663.
- Hasskarl, J. Everolimus. Methods Mol. Biol. 2018, 211, 101–123.
- Bachelot, T.; Bourgier, C.; Cropet, C.; Ray-Coquard, I.; Ferrero, J.-M.; Freyer, G.; Abadie-Lacourtoisie, S.; Eymard, J.-C.; Debled, M.; Spaëth, D.; et al. Randomized Phase II Trial of Everolimus in Combination With Tamoxifen in Patients With Hormone Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Metastatic Breast Cancer With Prior Exposure to Aromatase Inhibitors: A GINECO Study. J. Clin. Oncol. 2012, 30, 2718–2724.
- Royce, M.; Bachelot, T.; Villanueva, C.; Özgüroglu, M.; Azevedo, S.J.; Cruz, F.M.; Debled, M.; Hegg, R.; Toyama, T.; Falkson, C.; et al. Everolimus Plus Endocrine Therapy for Postmenopausal Women With Estrogen Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Advanced Breast Cancer. JAMA Oncol. 2018, 4, 977.
- Petrossian, K.; Nguyen, D.; Lo, C.; Kanaya, N.; Somlo, G.; Cui, Y.X.; Huang, C.-S.; Chen, S. Use of dual mTOR inhibitor MLN0128 against everolimus-resistant breast cancer. Breast Cancer Res. Treat. 2018, 170, 499–506.
- Lim, B.; Potter, D.A.; Salkeni, M.A.; Silverman, P.; Haddad, T.C.; Forget, F.; Awada, A.; Canon, J.-L.; Danso, M.; Lortholary, A.; et al. Sapanisertib Plus Exemestane or Fulvestrant in Women with Hormone Receptor–Positive/HER2-Negative Advanced or Metastatic Breast Cancer. Clin. Cancer Res. 2021, 27, 3329–3338.
- Schmid, P.; Zaiss, M.; Harper-Wynne, C.; Ferreira, M.; Dubey, S.; Chan, S.; Makris, A.; Nemsadze, G.; Brunt, A.M.; Kuemmel, S.; et al. Fulvestrant Plus Vistusertib vs Fulvestrant Plus Everolimus vs Fulvestrant Alone for Women With Hormone Receptor–Positive Metastatic Breast Cancer. JAMA Oncol. 2019, 5, 1556–1563.
- Brana, I.; Lorusso, P.; Baselga, J.; Heath, E.I.; Patnaik, A.; Gendreau, S.; Laird, A.; Papadopoulos, K. A phase I dose-escalation study of the safety, pharmacokinetics (PK), and pharmacodynamics of XL765 (SAR245409), a PI3K/TORC1/TORC2 inhibitor administered orally to patients (pts) with advanced malignancies. J. Clin. Oncol. 2010, 28, 3030.
- Funakoshi, T.; Latif, A.; Galsky, M.D. Risk of hematologic toxicities in patients with solid tumors treated with everolimus: A systematic review and meta-analysis. Crit. Rev. Oncol. 2013, 88, 30–41.
- Tabernero, J.; Rojo, F.; Calvo, E.; Burris, H.; Judson, I.; Hazell, K.; Martinelli, E.; Cajal, S.R.Y.; Jones, S.; Vidal, L.; et al. Dose- and Schedule-Dependent Inhibition of the Mammalian Target of Rapamycin Pathway With Everolimus: A Phase I Tumor Pharmacodynamic Study in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2008, 26, 1603–1610.
- Walker, E.H.; Perisic, O.; Ried, C.; Stephens, L.; Williams, R. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nat. Cell Biol. 1999, 402, 313–320.
- Yang, H.; Rudge, D.G.; Koos, J.; Vaidialingam, B.; Yang, H.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nat. Cell Biol. 2013, 497, 217–223.
- Hay, N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell 2005, 8, 179–183.
- O’Reilly, K.E.; Rojo, F.; She, Q.-B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR Inhibition Induces Upstream Receptor Tyrosine Kinase Signaling and Activates Akt. Cancer Res. 2006, 66, 1500–1508.
- Wainberg, Z.A.; Shapiro, G.; Curigliano, G.; Leong, S.; Kristeleit, R.S.; Maqueda, M.A.; Britten, C.D.; Milella, M.; Middleton, M.R.; Olszanski, A.J.; et al. Phase I study of the PI3K/mTOR inhibitor PF-05212384 in combination with other antitumor agents. J. Clin. Oncol. 2015, 33, 2590.
- Forero-Torres, A.; Han, H.; Dees, E.C.; Wesolowski, R.; Bardia, A.; Kabos, P.; Layman, R.M.; Lu, J.M.; Kern, K.A.; Perea, R.; et al. Phase Ib study of gedatolisib in combination with palbociclib and endocrine therapy (ET) in women with estrogen receptor (ER) positive (+) metastatic breast cancer (MBC) (B2151009). J. Clin. Oncol. 2018, 36, 1040.
- Franco, I.; Margaria, J.P.J.; De Santis, M.C.; A Ranghino, A.; Monteyne, D.; Chiaravalli, M.; Pema, M.M.; Campa, C.C.; E Ratto, E.; Gulluni, F.; et al. Phosphoinositide 3-Kinase-C2α Regulates Polycystin-2 Ciliary Entry and Protects against Kidney Cyst Formation. J. Am. Soc. Nephrol. 2015, 27, 1135–1144.
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383.
- Braccini, L.; Ciraolo, E.; Campa, C.C.; Perino, A.; Longo, D.L.; Tibolla, G.; Pregnolato, M.; Cao, Y.; Tassone, B.; Damilano, F.; et al. PI3K-C2γ is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signalling. Nat. Commun. 2015, 6, 7400.
- Yoshioka, K.; Yoshida, K.; Cui, H.; Wakayama, T.; Takuwa, N.; Okamoto, Y.; Du, W.; Qi, X.; Asanuma, K.; Sugihara, K.; et al. Endothelial PI3K-C2α, a class II PI3K, has an essential role in angiogenesis and vascular barrier function. Nat. Med. 2012, 18, 1560–1569.
- Valet, C.; Chicanne, G.; Severac, C.; Chaussade, C.; Whitehead, M.A.; Cabou, C.; Gratacap, M.-P.; Gaits-Iacovoni, F.; Vanhaesebroeck, B.; Payrastre, B.; et al. Essential role of class II PI3K-C2α in platelet membrane morphology. Blood 2015, 126, 1128–1137.
- Franco, I.; Gulluni, F.; Campa, C.C.; Costa, C.; Margaria, J.P.; Ciraolo, E.; Martini, M.; Monteyne, D.; De Luca, E.; Germena, G.; et al. PI3K Class II α Controls Spatially Restricted Endosomal PtdIns3P and Rab11 Activation to Promote Primary Cilium Function. Dev. Cell 2014, 28, 647–658.
- Marat, A.L.; Wallroth, A.; Lo, W.-T.; Müller, R.; Norata, G.D.; Falasca, M.; Schultz, C.; Haucke, V. mTORC1 activity repression by late endosomal phosphatidylinositol 3,4-bisphosphate. Science 2017, 356, 968–972.
- Campa, C.C.; Margaria, J.P.; Derle, A.; Del Giudice, M.; De Santis, M.C.; Gozzelino, L.; Copperi, F.; Bosia, C.; Hirsch, E. Rab11 activity and PtdIns(3)P turnover removes recycling cargo from endosomes. Nat. Chem. Biol. 2018, 14, 801–810.
- Ciraolo, E.; Gulluni, F.; Hirsch, E. Methods to Measure the Enzymatic Activity of PI3Ks. Methods Enzymol. 2014, 543, 115–140.
- Gozzelino, L.; De Santis, M.C.; Gulluni, F.; Hirsch, E.; Martini, M. PI(3,4)P2 Signaling in Cancer and Metabolism. Front. Oncol. 2020, 10, 360.
- Posor, Y.; Eichhorn-Gruenig, M.; Puchkov, D.; Schöneberg, J.; Ullrich, A.; Lampe, A.; Müller, R.; Zarbakhsh, S.; Gulluni, F.; Hirsch, E.; et al. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nat. Cell Biol. 2013, 499, 233–237.
- Wang, H.; Lo, W.-T.; Žagar, A.V.; Gulluni, F.; Lehmann, M.; Scapozza, L.; Haucke, V.; Vadas, O. Autoregulation of Class II Alpha PI3K Activity by Its Lipid-Binding PX-C2 Domain Module. Mol. Cell 2018, 71, 343–351.e4.
- Virbasius, J.V.; Guilherme, A.; Czech, M.P. Mouse p170 is a novel phosphatidylinositol 3-kinase containing a C2 domain. J. Biol. Chem. 1996, 271, 13304–13307.
- Domin, J.; Pages, F.; Volinia, S.; Rittenhouse, S.E.; Zvelebil, M.J.; Stein, R.C.; Waterfield, M.D. Cloning of a human phosphoinositide 3-kinase with a C2 domain that displays reduced sensitivity to the inhibitor wortmannin. Biochem. J. 1997, 326, 139–147.
- Martini, M.; Ciraolo, E.; Gulluni, F.; Hirsch, E. Targeting PI3K in Cancer: Any Good News? Front. Oncol. 2013, 3, 108.
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E.; Zhang, Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163.
- Gulluni, F.; Martini, M.; De Santis, M.C.; Campa, C.C.; Ghigo, A.; Margaria, J.P.; Ciraolo, E.; Franco, I.; Ala, U.; Annaratone, L.; et al. Mitotic Spindle Assembly and Genomic Stability in Breast Cancer Require PI3K-C2α Scaffolding Function. Cancer Cell 2017, 32, 444–459.e7.
- Chikh, A.; Ferro, R.; Abbott, J.J.; Piñeiro, R.; Buus, R.; Iezzi, M.; Ricci, F.; Bergamaschi, D.; Ostano, P.; Chiorino, G.; et al. Class II phosphoinositide 3-kinase C2β regulates a novel signaling pathway involved in breast cancer progression. Oncotarget 2016, 7, 18325–18345.
- Domin, J.; Harper, L.; Aubyn, D.; Wheeler, M.; Florey, O.; Haskard, D.; Yuan, M.; Zicha, D. The class II phosphoinositide 3-kinase PI3K-C2β regulates cell migration by a PtdIns(3)P dependent mechanism. J. Cell. Physiol. 2005, 205, 452–462.
- Katso, R.M.; Pardo, O.; Palamidessi, A.; Franz, C.M.; Marinov, M.; De Laurentiis, A.; Downward, J.; Scita, G.; Ridley, A.J.; Waterfield, M.D.; et al. Phosphoinositide 3-Kinase C2β Regulates Cytoskeletal Organization and Cell Migration via Rac-dependent Mechanisms. Mol. Biol. Cell 2006, 17, 3729–3744.
- Maffucci, T.; Cooke, F.T.; Foster, F.M.; Traer, C.J.; Fry, M.; Falasca, M. Class II phosphoinositide 3-kinase defines a novel signaling pathway in cell migration. J. Cell Biol. 2005, 169, 789–799.
- Falasca, M.; Hamilton, J.R.; Selvadurai, M.; Sundaram, K.; Adamska, A.; Thompson, P.E. Class II Phosphoinositide 3-Kinases as Novel Drug Targets. J. Med. Chem. 2016, 60, 47–65.
- Knight, Z.; Gonzalez, B.; Feldman, M.E.; Zunder, E.R.; Goldenberg, D.D.; Williams, O.; Loewith, R.; Stokoe, D.; Balla, A.; Toth, B.; et al. A Pharmacological Map of the PI3-K Family Defines a Role for p110α in Insulin Signaling. Cell 2006, 125, 733–747.
- Kong, D.; Dan, S.; Yamazaki, K.; Yamori, T. Inhibition profiles of phosphatidylinositol 3-kinase inhibitors against PI3K superfamily and human cancer cell line panel JFCR. Eur. J. Cancer 2010, 46, 1111–1121.
- Boller, D.; Doepfner, K.T.; De Laurentiis, A.; Guerreiro, A.S.; Marinov, M.; Shalaby, T.; Depledge, P.; Robson, A.; Saghir, N.; Hayakawa, M.; et al. Targeting PI3KC2beta impairs proliferation and survival in acute leukemia, brain tumours and neuroendocrine tumours. Anticancer Res 2012, 32, 3015–3027.
- Selvadurai, M.V.; Moon, M.J.; Mountford, S.J.; Ma, X.; Zheng, Z.; Jennings, I.G.; Setiabakti, N.M.; Iman, R.P.; Brazilek, R.J.; Abidin, N.A.Z.; et al. Disrupting the platelet internal membrane via PI3KC2α inhibition impairs thrombosis independently of canonical platelet activation. Sci. Transl. Med. 2020, 12, eaar8430.