Nuclear receptors (NRs) are ligand-dependent transcription factors that modulate diverse aspects of development, reproduction, and energy homeostasis. This receptor superfamily includes receptors for vitamin D, steroid hormones, thyroid hormones and retinoids, as well as a large number of orphan receptors. NRs are composed of six functionally distinct regions (termed A to F). The N-terminal AB region is highly variable and contains a constitutionally active transactivation function-1 (AF-1) motif. The central C region (a DNA-binding region) is highly conserved among NRs and contains two zinc finger motifs that make contact with specific nucleotide sequences, termed hormone response elements. The C-terminal D, E and F regions are required for ligand binding and receptor dimerization. In most NRs, these regions also contain a second highly conserved transcriptional activation function-2 (AF-2) motif, which is important for ligand-dependent transcription.
1. Overview
Progress in understanding peroxisome proliferator-activated receptor (PPAR) subtypes as nuclear receptors that have pleiotropic effects on biological responses has enabled the exploration of new subtype-selective PPAR ligands. Such ligands are useful chemical biology/pharmacological tools to investigate the functions of PPARs and are also candidate drugs for the treatment of PPAR-mediated diseases, such as metabolic syndrome, inflammation and cancer.
2. Nuclear Receptors
Nuclear receptors (NRs) are ligand-dependent transcription factors that modulate diverse aspects of development, reproduction, and energy homeostasis. This receptor superfamily includes receptors for vitamin D, steroid hormones, thyroid hormones and retinoids, as well as a large number of orphan receptors. NRs are composed of six functionally distinct regions (termed A to F) (Figure 1A). The N-terminal AB region is highly variable and contains a constitutionally active transactivation function-1 (AF-1) motif. The central C region (a DNA-binding region) is highly conserved among NRs and contains two zinc finger motifs that make contact with specific nucleotide sequences, termed hormone response elements. The C-terminal D, E and F regions are required for ligand binding and receptor dimerization. In most NRs, these regions also contain a second highly conserved transcriptional activation function-2 (AF-2) motif, which is important for ligand-dependent transcription.
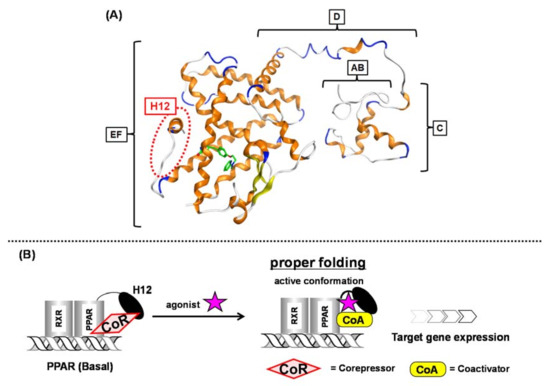
Figure 1. (A) Intact PPARγ–rosiglitazone complex structure. A to F regions are depicted. The position of H12 (helix 12) is indicated with a red dotted circle. (B) Schematic representation of ligand-dependent nuclear receptor activation (PPAR as an example). PPAR forms a heterodimer with another nuclear receptor, RXR in the nucleus.
In the basal state, NRs are functionally inactive because they are tightly bound with corepressors, such as NCoR and SMRT. Upon NR binding with an activating ligand, the corepressor dissociates and then coactivators, such as SRC-1 and NCoA, are recruited and the AF-2 helix located in the F region is stabilized to initiate transcription (
Figure 1B) [
1]. Over the past three decades, much attention has been focused on a subgroup of NRs, the peroxisome proliferator-activated receptors (PPARs).
3. Peroxisome Proliferator-Activated Receptors
PPARs are activated by endogenous unsaturated and saturated fatty acids and by synthetic ligands [
2]. There are three PPAR subtypes: PPARα, PPARδ, and PPARγ. Each PPAR subtype is expressed in a tissue-specific manner. PPARα is mostly expressed in tissues involved in lipid oxidation, such as liver and kidney. PPARγ is expressed in adipose tissue, macrophages and vascular smooth muscle, and also in tumors originating from various organs. PPAR
δ is expressed in adipose tissue, skeletal muscle, heart, etc. [
3].
Upon ligand binding, PPARs heterodimerize with another nuclear receptor, retinoid X receptor (RXR), in the nucleus, and the heterodimers regulate genes expression, such as carnitine palmitoyl acyl-CoA transferase 1A (
CPT1A), angiopoietin-like protein 4 (
ANGPTL4) and adipocyte differentiation-related protein (
ADRP) by binding to specific consensus DNA sequences, termed peroxisome proliferator responsive elements (PPREs) in the promoter regions of target genes. The structural basis of PPREs is a direct repeat of the hexameric AGGTCA recognition motif, separated by one nucleotide (termed DR1) [
4].
4. Pleiotropic Effect of PPARs
PPAR subtypes play a key role in lipid, lipoprotein and glucose homeostasis. PPARα regulates genes involved in fatty acid uptake, β-oxidation, and ω-oxidation. It downregulates apolipoprotein C-III, which regulates triglyceride hydrolysis by lipoprotein lipase, and also regulates genes involved in reverse cholesterol transport, such as apolipoprotein A-I and apolipoprotein A-II [
5]. PPARδ activation regulates HDL cholesterol levels, and it influences glycemic control [
6,
7,
8]. PPARδ activation markedly improves glucose tolerance and insulin resistance [
9]. PPARγ is a master regulator of adipocyte differentiation, but recent molecular studies have indicated that its activation is also linked to the expression of many important genes that affect energy metabolism, such as TNF-α, leptin, and adiponectin [
10]. PPARγ also promotes cell cycle arrest by inhibiting cyclin-dependent kinase activity in several tumor cell lines [
11].
Recent extensive biological studies clearly disclosed that PPARs function beyond metabolism. Each PPAR subtype plays major roles in a broad spectrum of biological processes, including cell proliferation and differentiation, fatty acid and eicosanoid signaling, bone formation, tissue repair and remodeling, insulin sensitivity [
12].
The above examples demonstrate that PPARs are important pleiotropic NRs and attractive molecular targets for the treatment of various diseases. Therefore, we think it is important to develop potent and PPAR subtype-selective ligands as tools to investigate the detailed functions of individual PPARs. In this review, we summarize in historical order our structural development studies to create PPAR subtype-selective agonists. The discovery of many types of PPAR ligands indicates the validity of our strategy to create subtype-selective NR agonists.
5. Working Hypothesis of the NR Ligand Superfamily
For over twenty years, we have been engaged in NR ligand structure research, which has been based on our working hypothesis of the NR ligand superfamily [
13]. The structural and functional features of the many different NRs are similar; therefore, we speculate that all NRs are derived from a single ancestral protein and have structurally evolved to fit various kinds of endogenous NR ligand. Similar evolution of NR ligands would have occurred from an ancestral ligand to form a superfamily of NR ligands, even though they now have diverse structures and functions. Based on this hypothesis, NR ligand structures are divided into two types, basic framework and branch. A common hydrophobic skeleton or a branch that fits into the ligand binding pocket of the basic ancestral protein are characteristic structural motifs that provide NR selectivity. PPAR subtype-selective agonists have certain unique structures associated with subtype selectivity. Examples include, thiazolidine-2,4-dione (TZD) and related structures, exemplified by pioglitazone, for PPARγ [
Figure 2 (1)], the 2,2-dialkyl(usually dimethyl)phenoxyacetic acid structure, exemplified by fenofibrate, for PPARα [
Figure 2 (2)], and the 2,2-unsubstituted phenoxyacetic acid structure, exemplified by GW-501516, for PPARδ [
Figure 2 (3)]. However, based on our working hypothesis, we predict that various kinds of subtype-selective, dual-, and pan-agonists can be created by starting with a common chemical framework as a template.
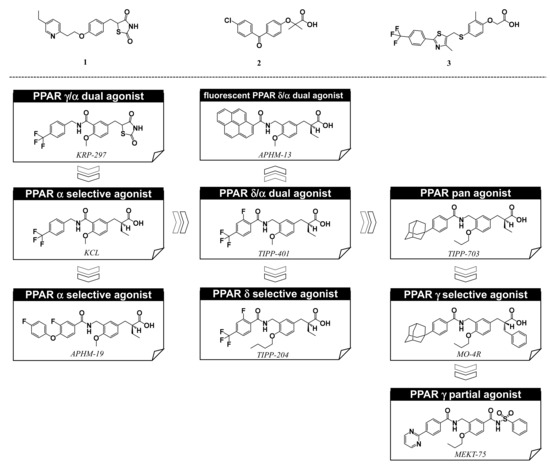
Figure 2. Structures of PPARγ agonist pioglitazone (1), PPARα agonist fenofibrate (2), and PPARδ agonist GW-501516 (3), and, below, our PPAR agonists.
Based on our hypothesis, we have successfully created various kinds of subtype-selective PPAR agonist, including KCL (PPAR α-selective agonist) [
14], APHM-19 (PPAR α-selective agonist) [
15], TIPP-401 (PPAR α/δ dual agonist) [
16], APHM-13 (fluorescent PPAR α/δ dual agonist) [
17], TIPP-204 (PPAR δ-selective agonist) [
18], TIPP-703 (PPAR pan agonist) [
19], MO-4R (PPAR γ-selective agonist) [
20], MEKT-21 (PPAR γ-selective partial agonist) [
21], and MEKT-75 (PPAR γ-selective partial agonist) [
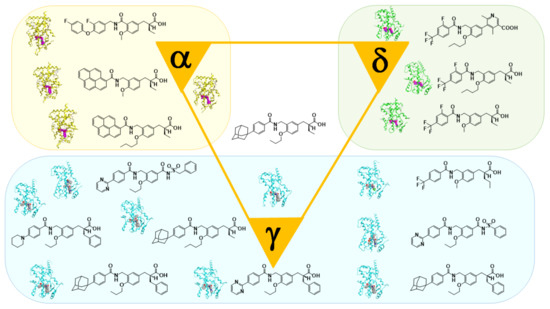
22]. In addition, in collaboration with Dr. Oyama (Yamanashi University, Japan) and Dr. Shimizu (Tokyo University, Japan), we have solved many X-ray crystallographic structures of PPAR subtypes complexed with our ligands [
23,
24,
25]. These structural biology studies have been integral to our medicinal chemistry research (
Figure 3).
Figure 3. X-ray-crystallographic structures of our representative PPAR agonists complexed with each PPAR ligand binding domain (LBD).
This entry is adapted from the peer-reviewed paper 10.3390/ijms22179223