Extracellular uric acid (UA) exhibits antioxidant properties by effectively scavenging free radicals in human plasma, but this benefit might be disturbed by the hydrophobic lipid layer of the cell membrane. In contrast, intracellular free oxygen radicals are produced during UA degradation, and superoxide is further enhanced by interacting with NADPH oxidase. This intracellular oxidative stress, together with inflammatory cytokines induced by UA, stimulates osteoclast bone resorption and inhibits osteoblast bone formation. UA also inhibits vitamin D production and thereby results in hyper-parathyroidism, which causes less UA excretion in the intestines and renal proximal tubules by inhibiting the urate transporter ATP-binding cassette subfamily G member 2 (ABCG2).
- uric acid
- osteoporosis
- oxidative stress
- inflammatory cytokines
- vitamin D deficiency
- secondary hyperparathyroidism
1. Introduction
2. The Clearance of Uric Acid (UA) in Humans
3. Osteoporosis
3.1. Normal Bone Remodeling
3.2. Coupling of Bone Stimulators and Turnover Inhibitors
3.3. The Pathogenesis of Osteoporosis
- (a) Increased inflammatory cytokine-associated osteolysis. It leads to excessive activity of osteoclasts and leads to more bone resorption than bone formation. This phenomenon can be seen in gouty arthritis, inflammation, and vitamin D deficiency [26].
- (b) Incoordination of the RANK/RANKL system. Increased RANK/RANKL signal in OC strengthens the performance of osteoclasts and brings about more bone resorption. There are many clinical scenarios such as in hyperparathyroidism and some autoimmune diseases such as rheumatoid arthritis, systemic lupus erythematosus, and estrogen deprivation (postmenopausal women) [26,27].
- (c) Excessive Wnt signaling inhibitors in osteoblasts. Dickkopf-1, sclerostin, and secreted frizzled related proteins lead to diminished functioning of osteoblasts via a decrease in Wnt signaling activity, then reduced bone formation. This has been proven in chronic kidney disease, glucocorticoid-induced osteoporosis, and vascular calcification-related bone loss [28].
4. The Uric Acid Oxidant and Antioxidant Paradox
4.1. Antioxidant Properties of Uric Acid in Human Plasma
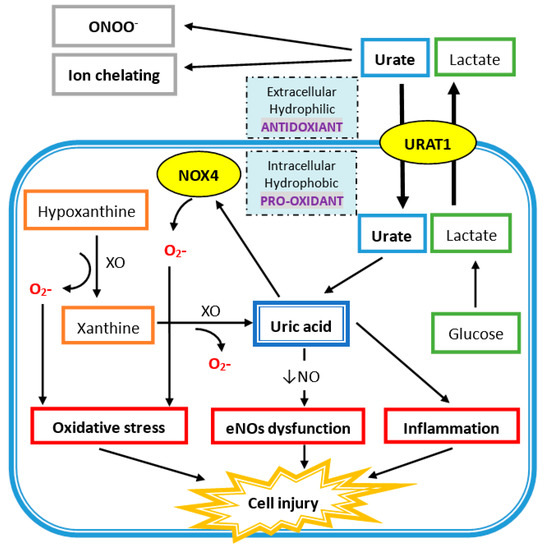
4.2. Intracellular Uric Acid Acts as a Pro-Oxidant to Damage Tissue
This entry is adapted from the peer-reviewed paper 10.3390/nu11092111