Microphthalmia-associated transcription factor (MITF) is a central player of melanocyte survival, function and development. It belongs to the MiT/TFE family of transcription factors in vertebrates, consisting of four distinct but closely related and evolutionary conserved members, including MITF, transcription factor EB (TFEB), TFE3 and TFEC. Structurally, MITF encodes a basic–helix–loop–helix leucine zipper (bHLH-ZIP) transcription factor, thereby exerting its function by regulating genes involved in cell cycle progression and differentiation, a role sustained throughout the process of melanogenesis and in melanoma.
- TPC
- two-pore
- lysosome
- TPC1
- TPC2
- TRPML
- MCOLN
- TRPML1
- MITF
- melanocytes
- melanoma
- mTOR
- TFEB
1. Introduction
Melanocytes are neural-crest derived cells that produce melanin, the primary determinant of skin color. Melanin is also found in hair, in the iris of the eye, and in the stria vascularis of the inner ear and, to a lesser degree, in a broad range of other tissues throughout the body [1]. There are two major types of melanin called eumelanin (dark, brown, black) and pheomelanin (yellow, red, light brown). Melanin is produced and stored in melanosomes, lysosome-related organelles that can be divided into four stages depending on their degree of maturation [2]. Stage I pre-melanosomes lack pigment but develop distinct fibrillar structures in a Pmel17-dependent process during stage II. Tyrosinase and other enzymes of melanogenesis reach stage II melanosomes via endosomal intermediates and initiate the production of melanin; these melanosomes are deposited on the fibers, resulting in their thickening and blackening with maturation to stage III. Melanin synthesis and deposition continue until all of the internal structure is masked in stage IV [3][4]. Modulators of melanin production include proteins involved in melanosome structure (i.e., Pmel17, MART-1), proteins involved in melanin synthesis and melanosome pH regulation (i.e., tyrosinase, TYRP1, TPC2), and proteins required for melanosome transport and distribution (e.g., Rab27a, myosin Va, melanophilin) [3][4]. Melanosomal pH is regulated by the vacuolar proton ATPase, Na + /H + exchangers, SLC24A5 (Na + /K + /Ca 2+ exchanger 5), and two-pore channel 2 (TPC2) [4][5][6][7][8]. TPC2, a cation channel permeable for sodium and calcium, expressed in late endosomes (LE), lysosomes and melanosomes, regulates pigmentation through two fundamental determinants of melanosome function: pH and size [7]. Different steps of melanogenesis are regulated by pH. First, the activity of tyrosinase, the rate-limiting enzyme for the production of melanin the optimal pH of which is 6.8 [9] with greatly reduced activities at a more acidic pH [5], while differences between monophenol oxidase and diphenol oxidase activities of tyrosinase in their pH dependence should be noted. Second, the activation of metatyrosinase (reduction of Cu II), the initial step of melanogenesis, is also highly pH-dependent. Third, the non-enzymatic DOPA-producing reaction, the redox exchange, is also strongly dependent on pH [10][11][12].
2. Role of MITF in Melanoma and Pathways Implicated
Microphthalmia-associated transcription factor (MITF) is a central player of melanocyte survival, function and development [13][14][15]. It belongs to the MiT/TFE family of transcription factors in vertebrates, consisting of four distinct but closely related and evolutionary conserved members, including MITF, transcription factor EB (TFEB), TFE3 and TFEC. Structurally, MITF encodes a basic–helix–loop– helix leucine zipper (bHLH-ZIP) transcription factor, thereby exerting its function by regulating genes involved in cell cycle progression and differentiation, a role sustained throughout the process of melanogenesis and in melanoma [16][17][18]. Mutations in MITF are associated with Tietz albinism-deafness syndrome and Waardenburg syndrome type 2A [19][20], and amplification of MITF is found in 15–20% of human metastatic melanomas and has been linked to poor survival. The M-MITF isoform is the predominant isoform in 80% of human melanomas [14][15][21].
MITF is regulated by numerous factors that exercise tight control on both its transcriptional and post-translational levels (i.e., ubiquitination, acetylation and sumoylation), given that it is downstream of several pathways [22][23], as illustrated in Figure 1 . A rheostat model of MITF in melanoma has been proposed by Carreira et al. (2006), where MITF yields three phenotypes, hence varied cellular responses in melanoma based on its activity. At the highest state where c-AMP induction leads to peak MITF activity, melanoma cells express differentiation target genes (i.e., Tyrosinase and MART1), giving rise to a pigmented phenotype and undergoing terminal differentiation. In contrast, the cells at the intermediate activity level are at a reversible proliferative state where enough MITF is expressed to activate MITF genes linked to survival (i.e., BCL2 and CDK2), concomitantly suppressing p27Kip1 expression through the regulation of Dia1, preventing differentiation. While MITF at its lowest activity brings about highly invasive cells exhibiting stem cell-like properties and low proliferative and pigmentation capacities that undergo p27Kip1-mediated G1 arrest [24][25]. Nonetheless, the rheostat model is controversial for many reasons. First, MITF is a downstream target of numerous signaling pathways and is subject to diverse post-translational modifications, which can direct MITF to different sets of target genes that regulate different functions in melanoma, dependent on physiological context [17][19]. Furthermore, findings that proliferative and invasive phenotypes are mutually exclusive has been disputed by Haass et al. (2014) [26].
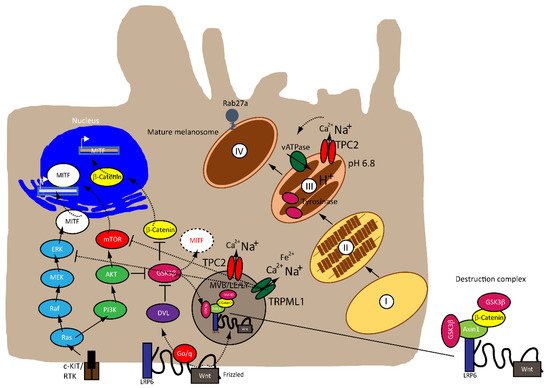
The mitogen-activated protein kinase (MAPK) pathway is dysregulated in 90% of melanoma with 50% of patients harboring activating mutations in B-RAF, most commonly the BRAF V600E mutation—an amino acid substitution from valine (V) to glutamic acid (E)—while 28% of patients carry mutations in N-RAS, leading to a constitutively hyperactivated MAPK signaling and increased melanoma cell survival, proliferation, migration, invasion, metastasis and angiogenesis. Consequently, the targeted therapies that are currently available have focused on developing specific small molecule tyrosine kinase inhibitors (TKI) for BRAF (BRAFi) and MEK (MEKi), which have shown improved survival rates [27][28][29][30][31].
The Wnt signaling pathway is divided into canonical β-catenin dependent and non-canonical β-catenin independent branches and is involved in regulating cellular homeostasis and development. Mutations of components of this pathway contribute to a number of pathologies, including familial exudative vitreoretinopathy, tooth development defects, Robinow syndrome, bone density defects, Alzheimer’s disease, and different types of cancers, such as melanoma, hepatocellular carcinoma (HCC), ovarian cancer, breast cancer and prostate cancer [32][33][34][35]. In melanoma, aberrations of the canonical Wnt pathway are directly linked to MITF and the rheostat model described above. The key player is β-catenin, which regulates genes of the melanocyte lineages and facilitates early-stage melanocyte transformation by blocking cellular senescence and increased proliferation. In melanoma cells, β-catenin activates proteins involved in melanogenesis and pigmentation such Melan-A, dopachrome tautomerase (DCT) and tyrosinase, all through MITF [34][36][37][38].
3. Endolysosomal Cation Channels in Melanoma
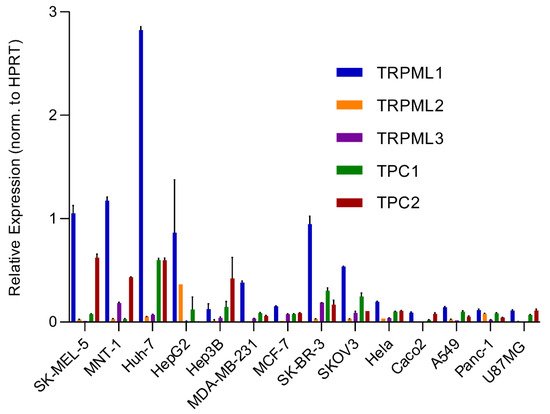
An analysis of the expression of different endolysosomal cation channels in a number of cancer cell lines using real-time quantitative reverse transcription PCR (qRT-PCR), including melanoma, hepatocellular carcinoma, breast cancer, colon adenocarcinoma, ovarian cancer, cervical adenocarcinoma, pancreatic ductal adenocarcinoma, glioblastoma and lung adenocarcinoma, is shown in Figure 2 .
Besides the involvement of MAPK and Wnt/ GSK3β/β-Catenin pathways described above, melanin formation is also triggered by melanocyte-stimulating hormone (MSH), a peptide hormone encoded by the proopiomelancortin gene (POMC). MSH binding to MC1R results in the induction of MITF via the cAMP response element-binding protein (CREB). In the TPC2 −/− MNT-1 cells, the expression levels of ERK, AKT, and CREB were found to be unchanged while the expression of GSK3β was increased [40]. It was thus concluded that the increased level of GSK3β results in reduced degradation of the GSK3β containing destruction complexes in endolysosomes, leading to increased GSK3β-dependent MITF degradation. These findings were recapitulated in MNT-1 cells treated with blockers for TPC2 [41] and it was found that flavonoid blockers of TPC2, such as naringenin [42], pratensein or duartin [41] like genetic loss of TPC2, increases melanin content and decreases proliferation, migration and invasion in a TPC2 dependent manner. In TPC2 −/− cells, no significant effects of the compounds were seen. In sum, Netcharoensirisuk et al. (2021) concluded that melanoma cell proliferation, migration and invasion are inversely correlated with TPC2-dependent melanin production in MNT-1 cells as the reduction of TPC2 expression increases melanin content but decreases proliferation, migration and invasion. It was suggested that this is the consequence of independent mechanisms: the regulation of MITF protein levels through interference with the endolysosomal activity of TPC2 and endolysosomal GSK3β degradation on the one hand and, on the other hand, the regulation of tyrosinase activity in melanosomes, independent of MITF by indirect interference with the melanosomal proton pump activity (decreased driving force for the pump due to absent or reduced TPC2 activity, resulting in reduced proton uptake by melanosomes, leading to less acidic melanosomal pH, increased tyrosinase activity, and eventually increased melanin production).
Clearly, this dual activity of TPC2 in melanosomes and endolysosomes is a special feature for TPC2 in melanoma cells, which requires further attention to understand its full potential as a possible drug target to treat melanoma.
This entry is adapted from the peer-reviewed paper 10.3390/biom11071021
References
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358.
- Yamaguchi, Y.; Hearing, V.J. Melanocytes and their diseases. Cold Spring Harb. Perspect. Med. 2014, 4, a017046.
- Raposo, G.; Marks, M.S. Melanosomes--dark organelles enlighten endosomal membrane transport. Nat. Rev. Mol. Cell Biol. 2007, 8, 786–797.
- Wasmeier, C.; Hume, A.N.; Bolasco, G.; Seabra, M.C. Melanosomes at a glance. J. Cell Sci. 2008, 121, 3995–3999.
- Ito, S.; Wakamatsu, K. Human hair melanins: What we have learned and have not learned from mouse coat color pigmentation. Pigment Cell Melanoma Res. 2011, 24, 63–74.
- Bellono, N.W.; Escobar, I.E.; Oancea, E. A melanosomal two-pore sodium channel regulates pigmentation. Sci. Rep. 2016, 6, 26570.
- Ambrosio, A.L.; Boyle, J.A.; Aradi, A.E.; Christian, K.A.; Di Pietro, S.M. TPC2 controls pigmentation by regulating melanosome pH and size. Proc. Natl. Acad. Sci. USA 2016, 113, 5622–5627.
- Chao, Y.K.; Schludi, V.; Chen, C.C.; Butz, E.; Nguyen, O.N.P.; Muller, M.; Kruger, J.; Kammerbauer, C.; Ben-Johny, M.; Vollmar, A.M.; et al. TPC2 polymorphisms associated with a hair pigmentation phenotype in humans result in gain of channel function by independent mechanisms. Proc. Natl. Acad. Sci. USA 2017, 114, E8595–E8602.
- Lerner, A.B.; Fitzpatrick, T.B.; Calkins, E.; Summerson, W.H. Mammalian tyrosinase; preparation and properties. J. Biol. Chem. 1949, 178, 185–195.
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 2004, 84, 1155–1228.
- Schallreuter, K.U.; Kothari, S.; Chavan, B.; Spencer, J.D. Regulation of melanogenesis--controversies and new concepts. Exp. Dermatol. 2008, 17, 395–404.
- Land, E.J.; Ramsden, C.A.; Riley, P.A. Quinone chemistry and melanogenesis. Methods Enzymol. 2004, 378, 88–109.
- Vachtenheim, J.; Borovanský, J. “Transcription physiology” of pigment formation in melanocytes: Central role of MITF. Exp. Dermatol. 2010, 19, 617–627.
- Levy, C.; Khaled, M.; Fisher, D.E. MITF: Master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006, 12, 406–414.
- Garraway, L.A.; Widlund, H.R.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, D.A.; Granter, S.R.; Du, J.; et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005, 436, 117–122.
- Steingrímsson, E.; Copeland, N.G.; Jenkins, N.A. Melanocytes and the microphthalmia transcription factor network. Annu. Rev. Genet. 2004, 38, 365–411.
- Slade, L.; Pulinilkunnil, T. The MiTF/TFE Family of Transcription Factors: Master Regulators of Organelle Signaling, Metabolism, and Stress Adaptation. Mol. Cancer Res. 2017, 15, 1637–1643.
- La Spina, M.; Contreras, P.S.; Rissone, A.; Meena, N.K.; Jeong, E.; Martina, J.A. MiT/TFE Family of Transcription Factors: An Evolutionary Perspective. Front. Cell Dev. Biol. 2021, 8, 1580.
- Ni, C.; Zhang, D.; Beyer, L.A.; Halsey, K.E.; Fukui, H.; Raphael, Y.; Dolan, D.F.; Hornyak, T.J. Hearing dysfunction in heterozygous Mitf(Mi-wh)/+ mice, a model for Waardenburg syndrome type 2 and Tietz syndrome. Pigment Cell Melanoma Res. 2013, 26, 78–87.
- Rauschendorf, M.A.; Zimmer, A.D.; Laut, A.; Demmer, P.; Rösler, B.; Happle, R.; Sartori, S.; Fischer, J. Homozygous intronic MITF mutation causes severe Waardenburg syndrome type 2A. Pigment Cell Melanoma Res. 2019, 32, 85–91.
- Wiedemann, G.M.; Aithal, C.; Kraechan, A.; Heise, C.; Cadilha, B.L.; Zhang, J.; Duewell, P.; Ballotti, R.; Endres, S.; Bertolotto, C.; et al. Microphthalmia-Associated Transcription Factor (MITF) Regulates Immune Cell Migration into Melanoma. Transl. Oncol. 2019, 12, 350–360.
- Cheli, Y.; Ohanna, M.; Ballotti, R.; Bertolotto, C. Fifteen-year quest for microphthalmia-associated transcription factor target genes. Pigment Cell Melanoma Res. 2010, 23, 27–40.
- Wellbrock, C.; Arozarena, I. Microphthalmia-associated transcription factor in melanoma development and MAP-kinase pathway targeted therapy. Pigment Cell Melanoma Res. 2015, 28, 390–406.
- Carreira, S.; Goodall, J.; Denat, L.; Rodriguez, M.; Nuciforo, P.; Hoek, K.S.; Testori, A.; Larue, L.; Goding, C.R. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes. Dev. 2006, 20, 3426–3439.
- Hoek, K.S.; Goding, C.R. Cancer stem cells versus phenotype-switching in melanoma. Pigment Cell Melanoma Res. 2010, 23, 746–759.
- Miller, A.J.; Levy, C.; Davis, I.J.; Razin, E.; Fisher, D.E. Sumoylation of MITF and its related family members TFE3 and TFEB. J. Biol. Chem. 2005, 280, 146–155.
- Barysch, M.J.; Mangana, J.; Dummer, R. A new B-Raf inhibitor combo for advanced melanoma. Oncotarget 2018, 9, 34457–34458.
- Greaves, W.O.; Verma, S.; Patel, K.P.; Davies, M.A.; Barkoh, B.A.; Galbincea, J.M.; Yao, H.; Lazar, A.J.; Aldape, K.D.; Medeiros, L.J.; et al. Frequency and spectrum of BRAF mutations in a retrospective, single-institution study of 1112 cases of melanoma. J. Mol. Diagn. 2013, 15, 220–226.
- Heppt, M.V.; Siepmann, T.; Engel, J.; Schubert-Fritschle, G.; Eckel, R.; Mirlach, L.; Kirchner, T.; Jung, A.; Gesierich, A.; Ruzicka, T.; et al. Prognostic significance of BRAF and NRAS mutations in melanoma: A German study from routine care. BMC Cancer 2017, 17, 536.
- Zhou, A.Y.; Johnson, D.B. Combinatorial Therapies in Melanoma: MAPK Inhibitors and Beyond. Am. J. Clin. Dermatol. 2018, 19, 181–193.
- Inamdar, G.S.; Madhunapantula, S.V.; Robertson, G.P. Targeting the MAPK pathway in melanoma: Why some approaches succeed and other fail. Biochem. Pharmacol. 2010, 80, 624–637.
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999.
- Gajos-Michniewicz, A.; Czyz, M. WNT Signaling in Melanoma. Int. J. Mol. Sci. 2020, 21, 4852.
- Johnson, M.L.; Rajamannan, N. Diseases of Wnt signaling. Rev. Endocr. Metab. Disord. 2006, 7, 41–49.
- De Ferrari, G.V.; Inestrosa, N.C. Wnt signaling function in Alzheimer’s disease. Brain Res. Brain Res. Rev. 2000, 33, 1–12.
- Larue, L.; Beermann, F. Cutaneous melanoma in genetically modified animals. Pigment Cell Res. 2007, 20, 485–497.
- Larue, L.; Luciani, F.; Kumasaka, M.; Champeval, D.; Demirkan, N.; Bonaventure, J.; Delmas, V. Bypassing melanocyte senescence by beta-catenin: A novel way to promote melanoma. Pathol. Biol. 2009, 57, 543–547.
- Delmas, V.; Beermann, F.; Martinozzi, S.; Carreira, S.; Ackermann, J.; Kumasaka, M.; Denat, L.; Goodall, J.; Luciani, F.; Viros, A.; et al. Beta-catenin induces immortalization of melanocytes by suppressing p16INK4a expression and cooperates with N-Ras in melanoma development. Genes. Dev. 2007, 21, 2923–2935.
- Pafumi, I.; Festa, M.; Papacci, F.; Lagostena, L.; Giunta, C.; Gutla, V.; Cornara, L.; Favia, A.; Palombi, F.; Gambale, F.; et al. Naringenin Impairs Two-Pore Channel 2 Activity And Inhibits VEGF-Induced Angiogenesis. Sci. Rep. 2017, 7, 5121.
- Grimm, C.; Bartel, K.; Vollmar, A.M.; Biel, M. Endolysosomal Cation Channels and Cancer-A Link with Great Potential. Pharmaceuticals 2018, 11, 4.
- Netcharoensirisuk, P.; Abrahamian, C.; Tang, R.; Chen, C.C.; Rosato, A.S.; Beyers, W.; Chao, Y.K.; Filippini, A.; Di Pietro, S.; Bartel, K.; et al. Flavonoids increase melanin production and reduce proliferation, migration and invasion of melanoma cells by blocking endolysosomal/melanosomal TPC2. Sci. Rep. 2021, 11, 8515.
- Sterea, A.M.; Almasi, S.; El Hiani, Y. The hidden potential of lysosomal ion channels: A new era of oncogenes. Cell Calcium 2018, 72, 91–103.