Stem cells have the capacity of self-renewal and, through proliferation and differentiation, are responsible for the embryonic development, postnatal development, and the regeneration of tissues in the adult organism. Cancer stem cells, analogous to the physiological stem cells, have the capacity of self-renewal and may account for growth and recurrence of tumors. Development and regeneration of healthy tissues and tumors depend on the balance of different genomic and nongenomic signaling pathways that regulate stem cell quiescence, proliferation, and differentiation. During evolution, this balance became dependent on all-trans retinoic acid (RA), a molecule derived from the environmental factor vitamin A.
1. Introduction
Retinoic acid (RA) regulates a wide range of biological processes during development and in adult organisms [
1,
2,
3,
4,
5,
6,
7,
8,
9]. Retinoic acid signaling is dependent on cells that can metabolize vitamin A (retinol) to RA. Retinol dehydrogenases oxidize retinol to retinal, and aldehyde dehydrogenases (ALDH1A1, ALDH1A2, and ALDH1A3) oxidize retinal to RA [
2,
10]. Retinoic acid released from these cells generates gradients that regulate neighboring cells. The precise RA level depends on the availability of vitamin A (retinol), the activity of enzymes involved in RA biosynthesis (retinol dehydrogenases and aldehyde dehydrogenases), and the RA catabolism by CYP26 enzymes [
11,
12,
13].
Retinoic acid regulates transcription by interacting with heterodimers of nuclear RA receptors (RARα, RARβ, and RARγ) and retinoid X receptors (RXRα, RXRβ, and RXRγ) bound to RA response elements (RAREs) in the promoters of target genes [
4,
14,
15]. The expression of over 500 genes is upregulated or downregulated by RA [
16]. Moreover, RA controls other transcriptional signaling pathways via different nuclear receptors, such as the peroxisome proliferator-activated receptor β/δ [
17,
18], and can also regulate different protein kinases in a nontranscriptional fashion [
19,
20,
21].
Although RA has been widely described as an inductor of cell differentiation, depending on cell-type, RA can antagonize cell differentiation and promote stemness (Table 1).
Table 1. Induction of stemness or cell differentiation by retinoic acid (RA) in a cell-type-dependent manner.
| Cell Type |
Action |
Signaling Pathway |
RA Dose-Time |
References |
| Pluripotent stem cells |
Stemness |
Inhibition of Wnt. Activation of Akt-mTOR |
0.5 µM (24 h) |
[22] |
| Breast cancer cells T47D403 |
Stemness |
Lack of expression of RARα tumor suppressor genes and activation of RARα-PI3K-AKT |
1 µM (72 h) |
[23] |
| Breast cancer cells MDA-MB-231 |
Stemness |
Upregulation of 1286 genes, among them MUC4.
Activation of the axis Src-YAP-IL6 |
0.1 µM (18 h)
5 µM (48 h) |
[24]
[25] |
| Breast cancer cells MDA-MB-468 |
Differentiation |
Upregulation of 1358 genes, among them HOXA1
Inhibition of the axis Src-YAP-IL6 |
0.1 µM (18 h)
5 µM (48 h) |
[24]
[25] |
| Breast cancer cells MCF-7 |
Stemness |
Activation of ALDH1A1-HIF1α-VEGF |
1 µM (48 h) |
[26] |
| Mammary MCF12A cells and T47D breast cancer cells |
Differentiation |
RARβ/TET2-miR200c-Suppression of PKCζ |
1 µM (24 h) |
[27] |
| Adult hippocampus |
Stemness |
Activation of HIF1α-VEGF |
1 µM (24 h) |
[28] |
| Glioblastoma T1440, T1452 and T1464 |
Stemness |
Increased SOX2 expression |
1 µM (7d) |
[29] |
| Glioblastoma T1338 |
Differentiation |
Decreased SOX2 expression |
1 µM (7d) |
[29] |
| Dormant hematopoietic cells |
Stemness |
Attenuation of C-MYC expression |
5 µM (24–48 h) |
[30] |
| Hematopoietic stem cells |
Differentiation or stemness |
Differentiation through RARα Stemness through RARγ NOTCH1 expression |
1 µM (14d) |
[31] |
| Colorectal cancer cells |
Differentiation |
RARγ-inhibition of YAP-increased E-cadherin expression |
1 µM (30 min) |
[32] |
| Hepatocelular carcinoma cells |
Stemness |
RARγ-PI3K-AKT-NFκB |
1 µM (48 h) |
[33] |
| Pancreatic ductal adenocarcinoma |
Differentiation |
Decrease ALDH1, SOX2 and NANOG |
10 μM (48 h) |
[34] |
| Spermatogonial stem cells |
Differentiation |
Upregulation of STRA8, AGPAT3, FAM57A, WDR91 |
0.1 μM (24 h) |
[35] |
| Regeneration of keratinocytes |
Stemness |
TLR3-STAT3 and NFkB-ALDH1-RA-RAR |
0.1µM (48 h) |
[36] |
2. Retinoic Acid Induces Stemness or Differentiation in the Mammary Gland and Breast Cancer Cells
Unlike other organs, the mammary gland tissue undergoes development postnatally. An adequate balance between stem self-renewal and stem cell differentiation is essential for this process. Prodifferentiation and antidifferentiation effects of RA have been reported during mammary gland development and breast cancer [
23,
24,
37].
2.1. Growth-Promoting and Growth-Inhibiting Actions of RA in Breast Cancer Depend on the Cell Context-Specific Balance of Activation of Transcriptional and Nontranscriptional Pathways
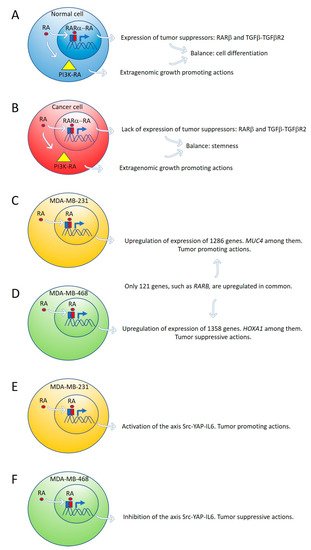
By global gene expression microarray analysis, Rossetti et al. [
23] determined that in breast cancer cells (T47D
Ctrl) grown under “physiological” RA culture conditions, many RARα-target genes, coding for tumor suppressor signaling pathways, as RARβ and the TGFβ-TGFβR2, are in a repressed transcriptional state marked by epigenetic histone modifications. In this situation, lack of expression of tumor suppressor genes cannot counteract the growth-promoting activity of nontranscriptional signaling pathways such as PI3K-AKT, triggered by direct interaction of RARα and the catalytic subunit of PI3K [
23]. The degree of inhibition of RARα transcriptional function is variable in different breast cancer cell lines: mild in T47D
Ctrl, severe in T47D
G303E, and extremely severe in T47D
403. Both in vitro and in vivo treatment with supraphysiological doses of exogenous RA significantly promoted T47D
403 breast cancer cell invasion [
23] (
Figure 1A,B).
Figure 1. Expression of tumor suppressor genes by retinoic acid (RA) in healthy cells counteracts the growth-promoting activity of nontranscriptional RA signaling pathways, such as PI3K-AKT (A). In cancer cells, lack of expression of tumor suppressor genes by RA cannot counteract the extragenomic tumor-promoting actions of RA (B). Differential gene expression induced by ALDH1A3 or RA in MDA-MB-231 and MDA-MB-468 cells (C,D). Retinoic acid upregulates the signaling pathway Src-YAP-IL6 involved in stemness in triple-negative MDA-MB-231 breast cancer cells (E) and downregulates the same pathway in triple-negative MDA-MB-468 breast cancer cells (F).
2.2. Retinoic Acid Induces Tumor-Promoting or Tumor-Suppressive Actions in Triple-Negative Breast Cancer Cells Due to Variable Gene Expression in Cell Lines with Differences in DNA Methylation
Marcato et al. [
24] reported that the effects of RA and ALDH1A3 activity were tumor-promoting in MDA-MB-231 and MDA-MB-435 triple-negative breast cancer cells, but tumor-suppressive in triple-negative MDA-MB-468 breast cancer cells. The opposing tumor growth effects of ALDH1A3/RA in breast cancer cells depend upon differential gene expression induced by ALDH1A3 or RA in MDA-MB-231 and MDA-MB-468 cells. Increased ALDH1A3 expression upregulated 1286 and 1358 genes in MDA-MB-231 and MDA-MB-468 cells, respectively. A large divergence in gene expression changes induced by ALDH1A3 in the two cell lines was observed because only 121 genes were upregulated in common in both cell lines.
RARβ is one of these genes (
Figure 1C,D).
One of the ALDH1A3-induced genes in MDA-MB-468 cells is the homeobox transcription factor A1 (HOXA1). The promotor of
HOXA1 possesses a RARE sequence that was previously shown to be inducible by RA [
38]. HOXA1 expression is significantly reduced by ALDH1A3 knockdown and induced by RA in MDA-MB-468 cells but is undetectable in MDA-MB-231 cells [
24].
HOXA1 is hypermethylated in MDA-MB-231 cells and hypomethylated in MDA-MB-468 cells [
24].
HOXA1 is often hypermethylated in cancer, suggesting a tumor-suppressive function [
39,
40].
Mucin 4 (
MUC4), a potential oncogene with a RARE, inducible by RA, and associated with triple-negative breast cancer [
41,
42], is significantly induced by ALDH1A3 and RA in MDA-MB-231 cells, but not in MDA-MB-468 cells.
MUC4 is hypermethylated in MDA-MB-468 and hypomethylated in MDA-MB-231 [
24]. MUC4 is typically hypomethylated in cancers, and its expression is associated with more aggressive cancer [
41,
42,
43,
44,
45].
MUC4 knockdown in MDA-MB-231 cells reduced their tumorigenic and metastatic properties [
42], suggesting
MUC4 may represent a gene that contributes to ALDH1A3/RA-mediated tumor growth and metastasis of MDA-MB-231 cells [
24].
2.3. Retinoic Acid Upregulates the Signaling Pathway Src-YAP-IL6 Involved in Stemness in Triple-Negative MDA-MB-231 Breast Cancer Cells and Downregulates the Same Pathway in Triple-Negative MDA-MB-468 Breast Cancer Cell Line
Retinoic acid induces tumor suppression in tumor xenografts of MDA-MB-468 breast cancer cells while increasing tumor growth and metastasis in xenografts of MDA-MB-231 [
24]. We have used these triple-negative breast cancer cell lines as a research model to investigate the role of RA on the regulation of the signaling pathway Src-YAP-Interleukin 6 involved in stemness [
25]. We found that RA activates this pro-invasive axis in triple-negative MDA-MB-231 breast cancer cells, yielding to an increased invasion of these cells. On the contrary, RA inhibits the Src-YAP-IL6 axis of triple-negative MDA-MB-468 cells, which results in decreased invasion phenotype (
Figure 1E,F). In both types of cells, inhibition of the Src-YAP-IL6 axis by the Src inhibitor PP2 drastically reduces migration and invasion. The Src-YAP-IL6 axis controls invasion, metastasis, resistance to therapy, and stemness of MDA-MB-231 breast cancer cells [
46,
47]. IL-6 is the first universal transcriptional target of YAP involved in promoting stemness conserved from flies to humans [
46,
48].
Overexpression of IL-6 induces cancer cell proliferation, angiogenesis, and metastasis through stimulating STAT3, MAPK, and Akt signaling pathways [
49]. IL-6 regulates cancer stem cell, mesenchymal stem cell formation, and epithelial to mesenchymal transition in cancer, and is a contributing factor for chemoresistance [
49]. Sansone et al. [
50] found that IL-6 mRNA was robustly elevated in mammospheres compared with breast epithelium and was required for their self-renewal and aggressive potential. Autocrine IL6-STAT3 signaling increases stem cell properties with efficient tumor colonization and outgrowth in vivo. Conversely, blockage of IL-6 reduces tumor burden and metastasis [
51,
52,
53,
54].
Nuclear YAP phosphorylation in MDA-MB-231 breast cancer cells depends on Src activity. Until recently, activation of YAP was believed to solely depend on the inhibition of the Hippo signaling pathway that retains YAP in the cytoplasm [
55]. To assess if YAP activation in MDA-MB-231 breast cancer cells depends on Src activity, as observed in other cancer cells [
56,
57,
58], we used Src inhibition by PP2, Src interference by siRNA and transfection of Src into MDA-MB-231 breast cancer cells. Src inhibition by PP2 and Src interference decreased YAP activity and downregulated IL-6 expression, while Src transfection activated YAP and upregulated IL-6 [
25].
The mechanism of Src activation induced by RA is not known at present. Mechanisms independent of transcription have been reported in breast cancer cells [
23]. However, the activation of the Src-YAP-IL6 axis we have observed should be the consequence of a genomic action of RA, given the 48 h delay following incubation with supraphysiological concentrations of RA (5 μM). Extragenomic effects of RA in breast cancer cells are produced faster and with lower levels of RA [
23].
Overexpression of MUC4 in triple-negative breast cancer cells induced by RA [
24] is an attractive candidate for Src activation because cell knockdown of MUC4 in pancreatic carcinoma decreased Src tyrosine phosphorylation significantly [
59]. IL-6 induces MUC4 expression through the gp130-STAT3 pathway in gastric cancer cell lines [
60].
An association of YAP activity and RA signaling with an increase in migration also has been observed in human neural crest cells [
61]. YAP, as well as its paralog TAZ, is known to act as a stemness-promoting factor in several tissue types, including hepatic, intestinal, and skin stem cell niches [
62,
63,
64,
65].
It has been reported that MDA-MB-231 and MDA-MB-468 are non-sphere-forming cells lines [
66]. However, it is not known how the presence of RA could affect mammosphere formation of these cell lines [
24] and whether these in vitro assays may reflect the expansion of breast cancer stem and nonstem cells in vivo. Using tumor xenografts, RA increases tumor growth and metastasis of MDA-MB-231 and decreases tumor growth of MDA-MB-468 cells [
24].
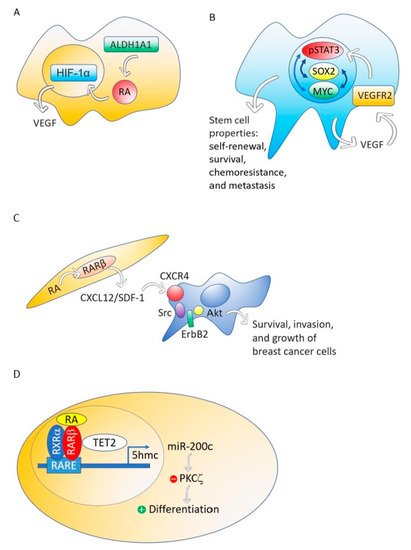
2.4. Retinoic Acid Conferred Stemness Properties to Breast Cancer MCF-7 Cells
Although different breast cancer cell lines such as 184A1, SUM149, SUM159, and HCC1954 treated with RA presented a decrease in mammosphere formation [
67], the breast cancer MCF-7 cell line responds to RA with an increase of stemness through an ALDH1A1-retinoic acid-HIF-1α-VEGF pathway [
26] (
Figure 2A). It has been reported that VEGF drives breast and lung cancer-initiating stem cells through the VEGFR-2-STAT3 signaling pathway that upregulates MYC and SOX2 [
68,
69] (
Figure 2B). VEGF contributes to the acquisition of stem cell properties, including self-renewal, survival, and chemoresistance through VEGFR2 receptors, VEGF neuropilin receptors [
70,
71] and intracrine VEGF receptors [
72,
73].
Figure 2. The ALDH1A1-retinoic acid-HIF-1α-VEGF pathway is activated in breast cancer MCF-7 cells (A). VEGF drives breast and lung cancer-initiating stem cells through the VEGFR-2-STAT3 signaling pathway that upregulates MYC and SOX2 gene expression (B). RA, through RARβ, increases the production of CXCL12/SDF-1 in stroma cells and, consequently, activates the Src-ErbB2-Akt signaling pathway in breast cancer cells, promoting survival, cell growth, and invasion (C). When the nontumorigenic, immortalized mammary epithelial cell line, MCF12A and the non-invasive breast cancer cell line T47D were treated with RA, the RA nuclear receptor RARβ associated with a methylcytosine dioxygenase (TET2) that produces DNA demethylation. The consequence is the induction of genes involved in cell differentiation and the activation of miR-200c expression. MiR-200c downregulates stemness targeting the protein kinase PKCζ. RA does not perform these actions in aggressive breast cancers (D).
2.5. RARβ Expression in the Mammary Gland Stroma Shapes the Tumor Microenvironment Favoring Breast Tumor Growth and Invasion
Although RARβ possesses, in breast cancer cells, many of the functional characteristics of a tumor suppressor, RARβ in the tumor stroma has a dominant role in promoting the growth and progression of mammary epithelial tumors [
74]. The mechanism through which stromal RARβ achieves its tumor-promoting effect probably involves the production of CXCL12/SDF-1 in stroma cells and the consequent activation of the Src-ErbB2-Akt signaling pathway in the breast cancer cells (
Figure 2C).
2.6. Retinoic Acid Induces Cell Differentiation and Downregulates Stemness in a Nontumorigenic Immortalized Mammary Epithelial Cell Line and a Non-Invasive Breast Cancer Cell line but Does Not Perform These Actions in Aggressive Breast Cancers
Using MCF12A, a nontumorigenic immortalized mammary epithelial cell line, or T47D, a non-invasive breast cancer cell line, RA induces genes involved in cell differentiation such as
RUNX1, BMP6, IKZF1 and
CAV1, and activates the expression of noncoding RNAs that downregulate stemness, such as miR-200c [
27]. This miRNA targets and suppresses the protein kinase PKCζ, a protein that has a pivotal role in directing the asymmetric division of mammalian stem cells to sustain the stem cell pool [
75,
76,
77]. PKCζ overexpression promotes breast cancer invasiveness and metastasis [
78]. However, the triple-negative breast cancer cell line MDA-MB-231 does not respond with cell differentiation and downregulation of stemness to RA treatment [
27].
Retinoic acid treatment of the nontumorigenic, immortalized mammary epithelial cell line, MCF12A and the non-invasive breast cancer cell line T47D induces the association of the RA nuclear receptor RARβ with a methylcytosine dioxygenase (TET2) [
27]. The TET protein family has a crucial role in DNA demethylation by catalyzing the conversion of the modified genomic base 5-methylcytosine into 5-hydroxymethylcytosine (5hmC), thereby activating the target gene expression [
79]. Expression of TET2 occurs in the nontumorigenic mammary epithelial cell line MCF12A and also in the non-invasive breast cancer cell line T47D, but repression occurs in aggressive breast cancers [
27] (
Figure 2D).
Retinoic acid enhanced the nuclear localization of RARβ and TET2, whereas knockdown of RARβ blocked RA mediated TET2 nuclear localization and substantially increased TET2 in the cytoplasm fraction. In contrast to nontumorigenic MCF12A and non-invasive breast cancer cell line T47D, TET2 was predominantly localized in the cytoplasm in aggressive triple-negative breast cancer cell line MDA-MB-231, which is deficient in endogenous RARβ expression. Re-expression of RARβ in MDA-MB-231 cells relocalized TET2 to the nucleus, and the nuclear TET2 level was further enhanced by RA treatment [
27].
2.7. Retinoic Acid Blocks the Progesterone Induction of Cytokeratin-5 Expressing Breast Cancer Stem Cells
Half of estrogen receptor-positive breast cancers contain a subpopulation of cytokeratin-5 expressing cells that are therapy-resistant and exhibit increased cancer stem cell properties induced by progesterone. Retinoic acid, through RARα or RARγ, blocks progesterone induction of cytokeratin-5 expression and stemness [
80].
This entry is adapted from the peer-reviewed paper 10.3390/biom9100567