Adult-onset Still’s disease (AOSD) is a non-familial, polygenic systemic autoinflammatory disorder. It is traditionally characterized by four cardinal manifestations—spiking fever, an evanescent salmon-pink maculopapular rash, arthralgia or arthritis and a white-blood-cell count (WBC) ≥ 10,000/mm3, mainly neutrophilic polymorphonuclear cells (PMNs)—but many other manifestations and complications can be associated, making clinical expression very heterogeneous and diagnosis sometimes difficult. The AOSD course can be diverse and is currently impossible to predict. Several clinical phenotypes have been described, either on the basis of the evolution of symptoms over time (monocyclic, polycyclic and chronic evolution) or according to dominant clinical evolution (systemic and arthritis subtypes). However, these patterns are mainly based on case series and not on robust epidemiological studies. Furthermore, they have mainly been established a long time ago, before the era of the biological treatments.
- adult-onset Still’s disease
- systemic-onset juvenile idiopathic arthritis
- spondyloarthritis
- psoriatic arthritis
- osteitis
- innate immunity
- autoinflammation
- immunological disease continuum
- phenotypes
- neutrophil urticarial dermatosis
1. Introduction
2. Common and Rare Clinical Manifestations
3. Evolution Courses
3.1. Classification Based on the Evolution Temporality
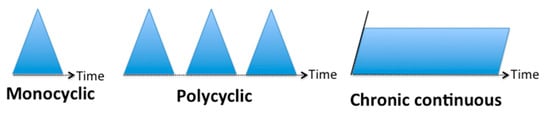
3.2. Classification Based on the Type of Symptoms
3.3. Pathogenic Consideration and Expected Therapeutic Outcomes
This entry is adapted from the peer-reviewed paper 10.3390/jcm10122633
References
- Feist, E.; Mitrovic, S.; Fautrel, B. Mechanisms, biomarkers and targets for adult-onset Still’s disease. Nat. Rev. Rheumatol. 2018, 14, 603–618.
- Fautrel, B. Adult-onset Still disease. Best Pract. Res. Clin. Rheumatol. 2008, 22, 773–792.
- Gerfaud-Valentin, M.; Jamilloux, Y.; Iwaz, J.; Sève, P. Adult-onset Still’s disease. Autoimmun. Rev. 2014, 13, 708–722.
- Yamaguchi, M.; Ohta, A.; Tsunematsu, T.; Kasukawa, R.; Mizushima, Y.; Kashiwagi, H.; Kashiwazaki, S.; Tanimoto, K.; Matsumoto, Y.; Ota, T. Preliminary criteria for classification of adult Still’s disease. J. Rheumatol. 1992, 19, 424–430.
- Fautrel, B.; Zing, E.; Golmard, J.-L.; Le Moel, G.; Bissery, A.; Rioux, C.; Rozenberg, S.; Piette, J.C.; Bourgeois, P. Proposal for a new set of classification criteria for adult-onset still disease. Medicine (Baltimore) 2002, 81, 194–200.
- Mitrovic, S.; Fautrel, B. New Markers for Adult-Onset Still’s Disease. Jt. Bone Spine 2018, 85, 285–293.
- Magadur-Joly, G.; Billaud, E.; Barrier, J.H.; Pennec, Y.L.; Masson, C.; Renou, P.; Prost, A. Epidemiology of adult Still’s disease: Estimate of the incidence by a retrospective study in west France. Ann. Rheum. Dis. 1995, 54, 587–590.
- Ohta, A.; Yamaguchi, M.; Tsunematsu, T.; Kasukawa, R.; Mizushima, H.; Kashiwagi, H.; Kashiwazaki, S.; Tanimoto, K.; Matsumoto, Y.; Akizuki, M. Adult Still’s disease: A multicenter survey of Japanese patients. J. Rheumatol. 1990, 17, 1058–1063.
- Pouchot, J.; Sampalis, J.S.; Beaudet, F.; Carette, S.; Décary, F.; Salusinsky-Sternbach, M.; Hill, R.O.; Gutkowski, A.N.; Harth, M.A.; Myhal, D.A. Adult Still’s disease: Manifestations, disease course, and outcome in 62 patients. Medicine (Baltimore) 1991, 70, 118–136.
- Masson, C.; Le Loët, X.; Lioté, F.; Renou, P.; Dubost, J.J.; Boissier, M.C.; Brithmer, L.; Bregeon, C.; Audran, M. Adult Still’s disease. Part II. Management, outcome, and prognostic factors. Rev. Rhum. Engl. Ed. 1995, 62, 758–765.
- Medsger, T.A.; Christy, W.C. Carpal arthritis with ankylosis in late onset still’s disease. Arthritis Rheum. 1976, 19, 232–242.
- Mitrovic, S.; Fautrel, B. Complications of adult-onset Still’s disease and their management. Expert Rev. Clin. Immunol. 2018, 14, 351–365.
- Lebrun, D.; Mestrallet, S.; Dehoux, M.; Golmard, J.L.; Granger, B.; Georgin-Lavialle, S.; Arnaud, L.; Grateau, G.; Pouchot, J.; Fautrel, B. Validation of the Fautrel classification criteria for adult-onset Still’s disease. Semin. Arthritis Rheum. 2018, 47, 578–585.
- Cush, J.J.; Medsger, T.A.; Christy, W.C.; Herbert, D.C.; Cooperstein, L.A. Adult-onset still’s disease. Arthritis Rheum. 1987, 30, 186–194.
- Bywaters, E.G. Still’s disease in the adult. Ann. Rheum. Diseases. 1971, 30, 121–133.
- Jamilloux, Y.; Gerfaud-Valentin, M.; Martinon, F.; Belot, A.; Henry, T.; Sève, P. Pathogenesis of adult-onset Still’s disease: New insights from the juvenile counterpart. Immunol. Res. 2015, 61, 53–62.
- Maria, A.T.J.; Le Quellec, A.; Jorgensen, C.; Touitou, I.; Rivière, S.; Guilpain, P. Adult onset Still’s disease (AOSD) in the era of biologic therapies: Dichotomous view for cytokine and clinical expressions. Autoimmun. Rev. 2014, 13, 1149–1159.
- Colafrancesco, S.; Priori, R.; Valesini, G. Presentation and diagnosis of adult-onset Still’s disease: The implications of current and emerging markers in overcoming the diagnostic challenge. Expert Rev. Clin. Immunol. 2015, 11, 749–761.
- Ichida, H.; Kawaguchi, Y.; Sugiura, T.; Takagi, K.; Katsumata, Y.; Gono, T.; Ota, Y.; Kataoka, S.; Kawasumi, H.; Yamanaka, H. Clinical Manifestations of Adult-Onset Still’s Disease Presenting With Erosive Arthritis: Association With Low Levels of Ferritin and Interleukin-18: Characteristics of AOSD With Severe Arthritis. Arthritis Care Res. 2014, 66, 642–646.
- Inoue, N.; Shimizu, M.; Tsunoda, S.; Kawano, M.; Matsumura, M.; Yachie, A. Cytokine profile in adult-onset Still’s disease: Comparison with systemic juvenile idiopathic arthritis. Clin. Immunol. 2016, 169, 8–13.
- Shimizu, M.; Yokoyama, T.; Yamada, K.; Kaneda, H.; Wada, H.; Wada, T.; Toma, T.; Ohta, K.; Kasahara, Y.; Yachie, A. Distinct cytokine profiles of systemic-onset juvenile idiopathic arthritis-associated macrophage activation syndrome with particular emphasis on the role of interleukin-18 in its pathogenesis. Rheumatology 2010, 49, 1645–1653.
- De Benedetti, F.; Brunner, H.I.; Ruperto, N.; Kenwright, A.; Wright, S.; Calvo, I.; Cuttica, R.; Ravelli, A.; Schneider, R.; Woo, P.; et al. Randomized Trial of Tocilizumab in Systemic Juvenile Idiopathic Arthritis. N. Engl. J. Med. 2012, 367, 2385–2395.