Metformin is a first-line treatment for many people with type 2 diabetes mellitus (T2DM) and gestational diabetes mellitus (GDM) to maintain glycaemic control. Although metformin demonstrates beneficial and protective cardiovascular effects for the mother, evidence suggests metformin may not be favourable for the short and long-term metabolic health of the offspring. Metformin can cross the placenta and could have a role in fetal programming. We postulate that a potential mechanism by which metformin may be adversely affecting placental and fetal development is by influencing maternal vitamin and micronutrient status.
- metformin
- diabetes
- placenta
- folate
- vitamin B12
- one carbon metabolism
- fetal growth
- LGA
- SGA
- fetal programming
1. Introduction
2. Metformin in Pregnancy
| Reference | Model | Effects Demonstrated by Metformin |
Significance |
|---|---|---|---|
| Clinical studies | |||
| Jamal et al. 2012 [32] |
Pregnant women with PCOS treated with metformin | - ⇔ on birth weight - ↓ uterine artery pulsatility index |
Metformin adversely affected uteroplacental circulation |
| Ex vivo or in vitro human placental studies | |||
| Jiang et al. 2020 [33] |
Human GDM and T2DM placental explants cultured and treated with metformin (ex vivo) | Male human placental explants: - AMPK activation - ↑ H3K27 acetylation - ↓ DNMT1 protein abundance - ↓ PGC-1α promoter methylation and ↑ PGC-1α mRNA expression |
Effects of metformin may be fetal sex-dependent Metformin may improve placental efficiency by facilitating placental mitochondrial biogenesis |
| Brownfoot et al. 2020 [34] Cluver et al. 2019 [35] Kaitu’u-Lino 2018 [36] Brownfoot et al. 2016 [37] |
Human primary tissues exposed to metformin; placental explants, endothelial cells and placental villous explants, whole maternal vessels, maternal omental vessel explants (in vitro and ex vivo) | - ↓ sFlt-1 and sEng secretion from primary endothelial cells, preterm preeclamptic placental villous explants and villous cytotrophoblast cells - ↓ VCAM-1 mRNA expression in endothelial cells - ↑ whole maternal blood vessel angiogenesis - ↓ sFlt mRNA expression - ↓ TNFα-mediated endothelial cell dysfunction |
Metformin enhances placental angiogenesis and reduces endothelial dysfunction by decreasing endothelial and trophoblastic antiangiogenic factor secretion via mitochondrial electron transport chain inhibition Metformin is being trialled as a medication for preeclampsia (trial number PACTR201608001752102) |
| Szukiewicz et al. 2018 [38] |
Human placental lobules perfused with metformin under normoglycemic or hyperglycaemic conditions (ex vivo) |
- ↓ CX3CL1 and TNFα secretion - ↑ placental CX3CR1 protein expression - ↓ placental NFκB p65 protein |
Metformin has anti-inflammatory effects in the placenta |
| Correia-Branco et al. 2018 [39] |
HTR-8/SVneo extravillous trophoblast cell line exposed to metformin (in vitro) |
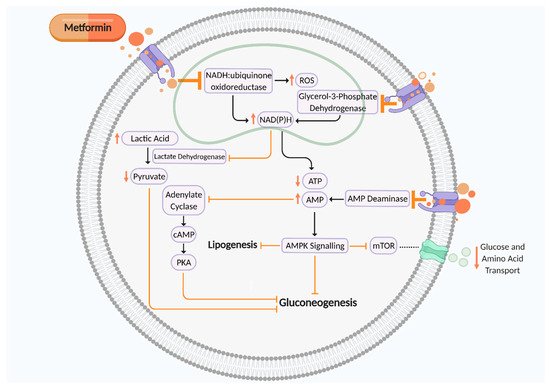
- ↓ proliferation - ↑ apoptosis - Inhibited folic acid uptake - Inhibited glucose uptake - Effects of metformin were prevented by inhibition of mTOR, JNK, and PI3K pathways |
Metformin impairs placental development and nutrient transport via PI3K, mTOR, JNK, and PI3K pathways |
| Arshad et al. 2016 [40] |
Human placental explants; from healthy pregnancy, non-treated diet-controlled GDM pregnancy, and metformin-treated GDM pregnancy (ex vivo) | - ↓ similar morphology in metformin-treated GDM placenta and non-treated healthy placenta, except for increased cord width - ↓ placental width in metformin-treated GDM placenta compared to non-treated GDM placenta - ↓ chorangiosis, placental thickness, and syncytial knots in metformin-treated placenta compared to non-treated GDM placenta |
Metformin may improve placental morphology by restoring diabetic placental hallmarks to characteristics similar to healthy placenta |
| Han et al. 2015 [41] |
Human first trimester trophoblasts treated with or without metformin (in vitro) | - ↓ trophoblast cytokine and chemokine release in normal and high glucose culture concentrations - No antiangiogenic or antimigratory effects |
Metformin may potentially decrease placental glucose-induced inflammatory response |
| In vivo rodent studies | |||
| Jiang et al. 2020 [33] |
Mice treated with maternal metformin and high-fat diet | Improved placental efficiency in males: - ↓ PGC-1α promoter methylation and ↑ PGC-1α expression - ↑ TFAM expression Improved glucose homeostasis in male offspring |
Metformin may improve placental efficiency by facilitating placental mitochondrial biogenesis Metformin may be protective to the offspring by suppressing epigenetic changes evoked by maternal diabetes |
| Wang et al. 2019 [42] |
Pregnant mice fed an isocaloric diet (control), high-fat diet, or high-fat diet plus metformin (in vivo) |
- ↓ placental weight compared to control - Partially rescued high-fat diet induced ↓ in placental and fetal weight - ↑ VEGF and MMP-2 protein expression |
Metformin improves high fat diet-induced reduction in placental and fetal growth, potentially by modulating placental vasculature |
| Alzamendi et al. 2012 [43] |
Pregnant rats fed a normal or high-fructose diet, treated with metformin (in vivo) |
- ↓ fetal weight - ⇔ on placental weight or blood vessel area - Improved fructose diet induced ↓ blood vessel area |
Metformin reduces fetal weight in mice fed a normal diet Metformin prevents high fructose diet-induced placental dysfunction |
Dark grey is table heading; pale grey titles demonstrate whether the study was clinical, ex-vivo or in vitro human placental, or in-vivo rodent studies. ⇔ no change; ↓reduction; ↑ increase. AMPK, AMP-activated protein kinase; DNMT, DNA methyltransferase; PGC-1α, peroxisome proliferator-activated receptor-gamma coactivator 1α; TFAM, mitochondrial transcription factor A; sFlt-1, soluble fms-like tyrosine kinase-1; sEng, soluble endoglin; VCAM-1, vascular cell adhesion molecule 1; TNFα, tumour necrosis factor alpha; VEGF, vascular endothelial growth factor; MMP-2, matrix metalloproteinase-2; NF-κB, nuclear factor kappa B; mTOR, mammalian target of rapamycin; JNK, c-Jun N-terminal kinase; PI3K, hosphatidylinositol-3-kinase.
2.1. Transplacental Transport of Metformin
2.2. Impact of Metformin on Placental Nutrient Transport and Nutrient Bioavailabilty
This entry is adapted from the peer-reviewed paper 10.3390/ijms22115759