1. Tumor Necrosis Factor (TNF) and Tumor Necrosis Factor-α Converting Enzyme (TACE)
TNF was described for the first time in the middle of 1970; it is a polypeptide considered a potent pro-inflammatory cytokine and encoded in the major histocompatibility complex in human and mice, and it is produced by immune system cells both myeloid and lymphoid origin [
1,
2,
3].
TNF is synthesized as a monomeric protein and stored in vesicles, which use the route of rough endoplasmic reticulum (ER) to cross the cytoplasm. TNF monomers form a compact trimer through non-covalent interactions; the TNF-trimeric has high thermodynamic stability, the molecular mass of the human TNF is 50.4-kDa and murine TNF 50-kDa [
4,
5]. Active TNF is also expressed as a trimeric transmembrane form on the cell surface (hereafter tmTNF); after an activation stimulus, tmTNF is proteolytically processed, and a soluble form is released (hereafter solTNF); both tmTNF and solTNF display physiological functions [
6].
TACE or also called A Disintegrin and Metalloproteinase (ADAM) domain 17 (ADAM17), cleaves tmTNF between residues Ala76 and Val77 to obtain solTNF [
7]. TACE is the only, or at least the major sheddase of TNF in vivo, other ADAM family members such as ADAM10, ADAM9, and ADAM19 have been shown to shed TNF only in vitro and the cleavage site does not match with the physiologically relevant site [
8]. TACE is not a TNF specific protease; several other proteins such as transforming growth factor-beta (TGF-β), beta-amyloid precursor protein, and TNF receptors are released by the TACE action [
8,
9]. It has been well documented that TACE also mediates the cleavage of the receptor Angiotensin-converting enzyme-2, a receptor used by viruses such as SARS-CoV and SARS-CoV2 to infect the cells [
10,
11,
12].
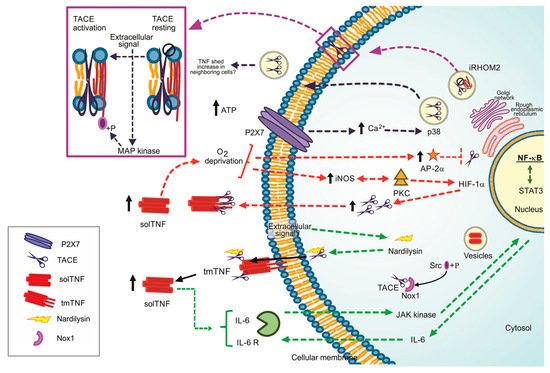
It is currently well established that TACE is necessary to deliver solTNF; however, how its catalytic activity is activated or regulated is still limited. Probably, the presence of a functional TACE is dependent on the action of multiple molecules. For instance, in TGF-β-mediated TACE activation, when TGF-β binds to its receptors, the sarcoma kinase (Src) molecule is phosphorylated, mediating NOX 1 activation and producing reactive oxygen species (ROS), which is finally activating TACE [
13]. The TNF-dependent pathway activates the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signalling, whereas the Uev1A-Ubc13 complex catalyzes the ubiquitination of RHBDF2, which is a crucial factor to promote TACE maturation. Consequently, Uev1A-Ubc13 inhibition interferes with RHBDF2-promoted TACE maturation [
14].
Three mechanisms proposed to explain how the TACE’ catalytic activity is regulated are discussed below.
(1) Nardilysin (NRD)-dependent pathway: NRD is a metalloendopeptidase of the M16 family. NRD binds to TACE (at cytosol or extracellular level) and potentiates its catalytic activity, a process promoted by phorbol esters. The NRD gene’s overexpression increases TNF and induces ADAM10 activation, suggesting that the high TNF shedding could be due to the function of both TACE and ADAM10 [
15,
16] (, dotted green line). Although the TNF shedding by ADAM10 has been observed only as an in-vitro process, using cultures of TACE-deficient fibroblasts, solTNF was detected, suggesting that ADAM10 is the TNF shedder when TACE is not present [
17].
Figure 1. Regulation pathways to the TNF/TACE axis, ways to lead the TACE activation. Dotted green line: Nardilysin-dependent pathways. Dotted red line: iNOS/AP-2a-dependent pathways. +P Phosphorylate form. Dotted blue black line: P2X7 dependent pathway, Dotted purple line: iRHOM2 as the TACE maturation promoter. Black arrow up: increased level.
TNF can promote cancer cells’ proliferation [
18]. NRD increases TNF shedding; consequently, NF-κB signalling pathway and pro-inflammatory microenvironment, characterized by the presence of interleukin (IL)-1β, IL-6 and prostaglandin E2 are activated. In turn, STAT3 is phosphorylated by the autocrine function of IL-6, and growth-related genes are upregulated [
19] (, dotted green line). Several questions remain open, for instance, if a specific extracellular signal is necessary or not to induce the complex NRD/TACE or if ADAM10 can or not cleave TNF in vivo.
(2) Oxygen deprivation-dependent pathway. Reports suggested that oxygen deprivation induces increased TACE expression and, consequently, high solTNF levels [
20]. By this pathway, the protein kinase C (PKC) and inducible nitric oxide synthase (iNOS) induce a nuclear accumulation of NF-κB and hypoxic-inducible factor-1 subunit α (HIF-1α) to stimulate TACE promotor activity and solTNF increase by an autocrine function, thus the shedding of TNF could be perpetuated [
21,
22] (, dotted red line).
Reports also suggested that solTNF upregulates the transcription factor AP-2α. However, the TACE promoter contains an AP-2α binding sequence, and it can bind in a TNF-dependent manner, indicating that solTNF downregulates TACE expression and function because AP-2α is enhanced [
23] (, dotted red line). The role played by AP-2α in the synthesis and function of TACE is controversial. Evidence showed that TNF induces caspase-6 activation, which in turn cleaves AP-2α, suggesting that TNF downregulates AP-2α by a caspase 6-dependent pathway [
24]. It is possible that the concentration of solTNF and tmTNF, or maybe one of the receptors, could be responsible for determining the up-or down-regulation of AP-2α and its consequent role.
(3) P2 and iRHOM2 pathways. The P2 purinergic receptors have bi-functional effects on TNF release. On the one hand, P2X receptor activation attenuates TNF release and simultaneously, on the other hand, P2Y induces TNF release [
25]. ATP induces intracellular Ca
2+ rise by P2X7-dependent pathway, activating kinase p38 and finally promoting TACE’s release into exosomes. It could be a mechanism to shed membrane proteins to neighboring cells, thus propagating inflammation [
26] (, dotted black line).
iRHOM2 (rhomboid 5 homolog 2) or RHBDF2 is a member of the rhomboid protein family found in the ER. iRHOM2 is considered an essential regulator for the crosstalk TNF/TACE as it helps TACE to get the plasma membrane [
27]. Data indicate that Ubiquitin (Ub)-conjugating enzyme variant 1A (Uev1A) polyubiquitinates iRHOM2 promoting TACE maturation (, dotted purple line). However, the role of iRHOM2 is not limited to induce TACE maturation. The phosphorylation of the iRHOM2 cytoplasmic tail by MAP kinases (p38, JNK and ERK1/2) is a crucial step to expose TACE proteolytic site activating its sheddase function [
14,
28] (, purple box).
2. Tumor Necrosis Factor Receptors
Two receptors have been identified to mediate interactions with TNF, tumor necrosis factor receptor 1 (TNFR1), also called CD120a and p55 (its molecular weight is 55 kDa), and tumor necrosis factor receptor 2 (TNFR2), also called CD120b and p75 (its molecular weight is 75 kDa) [
29]. TNFR1 and TNFR2 are not specific to TNF, they interact also with lymphotoxin alpha (LTα, previously known as TNFβ). LTα is a cytokine closely related to TNF, activated by similar stimuli than those activating TNF, produced mainly by lymphoid cells in a soluble form and can combine with LTβ interacting with another different receptor, LTβR [
30].
TNFR1 and TNFR2 are on the cellular membrane or in a soluble form following TACE activation; their cytoplasmic domains are unrelated, and intracellular signalling pathways are independent. TNFR1 is involved in cytotoxicity, whereas TNFR2 plays a role in cytotoxicity and proliferation [
31,
32]. As described in the previous section, the exact mechanism involving TACE in the shedding of TNFR1 and TNFR2 is still unclear.
2.1. Tumor Necrosis Factor Receptor 1
TNFR1 contains a cytoplasmic region designed death domain (DD), which initiates a signal of cytotoxicity with homology to the intracellular domain of Fas antigen [
33]. TNFR1 activates different signalling pathways, including neutrophil migration, complement pathway, regulation of other cytokines and chemokines, adhesion molecules and their receptors, generally promoting inflammatory responses [
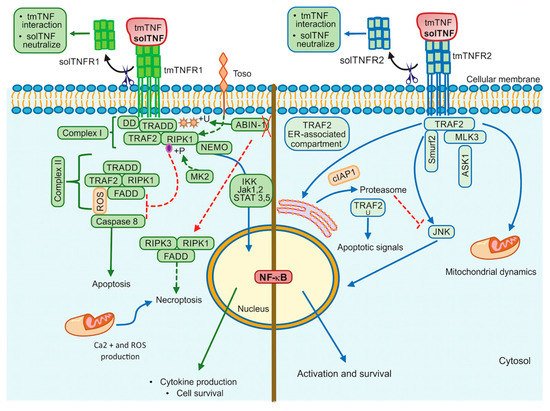
34]. Several molecules were described as essential for NF-κB activation through a TNFR1-dependent pathway, for example, TRADD (TNFR1-associated death domain), RIPK1 (receptor-interacting protein kinase 1, also known as RIP1), TRAF2 (TNFR-associated factor 2) and FADD (Fas-associated death domain) [
35]. The TNF/tmTNFR1 complex induces activation signalling pathways leading to opposite effects such as cell survival and cell death. Once TNF binds tmTNFR1, TRADD, RIPK1, TRAF2 and TAK1 molecules are recruited near DD and complex I. This complex I mediates the activation of MAPK and NF-κB, promoting cell survival. To activate this pathway, the phosphorylation of Jak-(Janus kinase)1 and Jak2, STAT-(signal transducer and activator of transcription)3 and STAT5 are required [
36,
37]. RIPK1 (RIPK1
U) is ubiquitinated to recruit Ikappa B kinase (IKK), and then there is a binding between RIPK1
U/NEMO (regulatory subunit IKKγ) to finally activate NF-κB [
38] (, left). However, if TRADD, RIPK1 and TRAF2 are dissociated from DD, they can interact with FADD, assembling the complex II that activates Caspase-8, inducing cell death. It has been suggested that to induce a full caspase 8-activation, ROS generation is required upstream and downstream of complex II [
37,
39] (, left).
Figure 2. TNFR1 and TNFR2 activation pathways. TNFR1 signalling is involved in cell death events and inflammatory processes by classical NF-kB, whereas TNRF2 triggers signalling that may activate classical or non-classical NF-kB pathway. Additionally, TNFR2 impacts mitochondrial dynamics. ABIN-1: Ubiquitin binding protein, DD: Death domain, FADD: Fas-associated death domain, IKK: IkappaB kinase, Jak: Janus kinase protein, NEMO: The NF-κB essential modulator (or IKKγ), RIPK1: Receptor interacting protein kinase 1, RIPK3: Receptor interacting protein kinase 3, ROS: Reactive oxygen species, STAT: Signal transducer and activator of transcription proteins, Toso: Human Fcμ receptor (hFCMR) or FAIM3, TRADD: TNFR1-associated death domain, TRAF2: TNFR-associated factor 2, +P: Phosphorylation, +U: Ubiquitination. Continuous lines green and blue: activation signalling. Dotted lines: inhibition signalling. A red cross: absence.
The interactions of solTNF or tmTNF with tmTNFR1 are also crucial to trigger apoptotic signals. It has been recently shown that tmTNF induces the binding of STAT1 to a region spanning amino acids 319–337 of tmTNFR1. STAT1-phosphorylation (at the serine residue in position 727) favors its binding to TRADD and FADD, promoting apoptosis but not NF-κB activation [
40]. Thus, the regulatory balance between survival versus death by the TNFR1 pathway is crucial to maintain homeostasis, and several authors suggested that TNFR1-mediated-signal transduction includes a checkpoint resulting in cell death when the signal activating NF-κB fails. Although it is not yet clarified how this checkpoint is controlled, experimental evidence suggested that RIPK1 is a crucial target to regulate this balance. It was reported that IKKα/IKKβ mediates direct phosphorylation of RIPK1 as the last step regulating cell death [
41].
Toso, also known as FAIM3 (Fas apoptosis inhibitory molecule 3) or FcμR, is a transmembrane protein with a negative regulatory function, which promotes the ubiquitination of RIP1 inducing NF-κB activation and consequently cell survival [
42], affecting caspase 8 activation [
43]. Another molecule that has been involved in cell death is MAPKAP kinase-2 (MK2) [
44]. If the effector MK2 induces phosphorylation of the kinase RIPK1, the binding with FADD/Caspase 8 is inhibited, thus complex-II-dependent cell death is blocked [
44,
45]. TNFR1-mediated cell survival may depend on ubiquitination and phosphorylation of RIPK1 (, left).
Additionally, TNF/tmTNFR1 pathway can induce cell death called necroptosis as the assembly of FADD, RIPK1, and RIPK3 form the complex called necrosome [
46]. Roca and collaborators [
47], have shown in zebrafish or in human macrophages (infected with
Mycobacterium marinum or
Mycobacterium tuberculosis) that an excess of TNF triggers necrosis through TNF-RIPK1-RIPK3 interactions increasing the production of ROS, cyclophilin D (mitochondrial matrix protein), BID and BAX (pro-apoptotic proteins) [
48]. The same group has recently reported that TNF triggers the production of ROS activating cyclophilin D (mitochondrial matrix protein) and leading to BID and BAX (pro-apoptotic proteins) activation, which results in mitochondrial Ca
2+ overload through ER ryanodine receptor and necrosis [
47].
The Ubiquitin-binding protein ABIN-1 is critical for the activation of RIPK1 [
49]. This protein is recruited into the complex I and plays a critical role in the control of ubiquitylation and deubiquitylation. It has been proposed that ABIN-1 deficiency reduces the recruitment of A20 molecule (negative regulator of NFκB), and consequently, there are ubiquitylation and activation of RIPK1 to mediate necroptosis [
49] (, left).
2.2. Tumor Necrosis Factor Receptor 2
Although the tmTNFR2 signalling pathway has been mainly implicated in cell proliferation, activation and survival, data have also reported to transduce apoptotic signal under specific models and potentiate TNFR1-induced cell death [
50,
51].
Two molecules with the ability to interact with the TNFR2 cytoplasmic domain and to induce signalling were reported and called TNF receptor-associated factor 1 (TRAF1) and TRAF2 [
52]. TNFR2 signalling was first simplified as a pathway where TRAF2 induces c-Jun-N-terminal kinase (JNK) activation, using the apoptosis signal-regulating kinase 1 (ASK1) as a mediator to activate NF-κB and facilitating anti-apoptotic signals [
53].
JNK plays a fundamental role to determine the outcome of the TNFR2 pathway. It has been shown that after TNF/tmTNFR2 engagement, TRAF2 and the inhibitor of apoptosis molecules called cIAP1 (molecule to mediate the TRAF2 ubiquitination) are recruited to the TNFR2 cytoplasmic domain, and later it is translocated to an ER-associated compartment, where TRAF2 ubiquitination occurs [
54,
55] (, right). The ER is sensitive to homeostasis alterations favoring the accumulation of misfolded proteins, which trigger ER stress and promote apoptotic cell death [
56]. Thus, after ER-stress, TRAF2 forms a complex with the ER-stress sensor called IRE1 and interacts with procaspase-12 promoting its activation [
57]. It has also been proposed that ER-stress induces expression of TNF in an IRE1- and NF-κB-dependent manner, and TRAF2 is decreased; these together inhibit the JNK activation and makes cells susceptible to cell death [
58]. Those data support evidence for a relevant role of the ER in an additional apoptotic control point of the TNFR2 pathway.
It has also been proposed that following tmTNFR2 activation, the E3 ubiquitin ligase Smurf2 forms a ternary complex with tmTNFR2 and TRAF2, inducing relocalization of TNFR2 to the insoluble membrane/cytoskeletal fraction promoting the JNK activation [
59]. Mixed lineage kinase 3 (MLK3), a mitogen-activated protein kinase kinase kinase (MAP3K) required for optimal activation of JNK signalling, has been shown to associate with TRAF2, TRAF5 and TRAF6. However, only TRAF2 induces the kinase activity of MLK3 by conjugating with polyubiquitin chains to activate JNK [
60,
61].
Finally, there is controversial evidence about the role of TNF on mitochondrial integrity. Reports have shown that TNF causes mitochondrial fragmentation (fission), but other authors suggested that TNF stimulates mitochondrial biogenesis [
62,
63]. In human airway smooth muscle (hASM) non-asthmatic cells, fission was associated with an increased level of Drp1 (Dynamin related protein (1) and the decreased level of Mfn2 (GTPase Mitofusin (2), and TNF was involved in Mfn2 reduction [
64]. Recently, it was reported that hASM cells exposed to TNF showed fission and mitochondrial biogenesis, with increased organelle volume density, cell proliferation, reduction of Ca
2+ influx, and decrease of O2 consumption per mitochondrion [
65]. In this regard, the TNFR2-activation mediates interactions between Stat3 (signal transducer and activator of transcription (3) and Re1A (Stat-Re1A protein) as the Stat3/Re1A complex interacts with two lysine residues within Stat3 that is acetylated by p300 and OPA1 (optic atrophy 1) expression is increased, suggesting an enhanced mitochondrial fusion [
66].
In summary, the tmTNFR2 pathway is tightly regulated depending on different cellular organelles, which are alternative for specific cellular systems when TNF is produced in inappropriate concentration. These mechanisms can help to develop novel therapeutic targets for treating or preventing diseases where TNF expression is dysregulated.
3. tmTNF Signalling Pathway
As previously discussed, tmTNF is a stable homotrimer that is cleaved by TACE and released as solTNF. The first publications on tmTNF suggested that tmTNF interacts primarily with TNFR2, whereas solTNF binds mainly to TNFR1 [
67,
68]. However, we need to consider that the tmTNF form that showed the specific interaction with TNFR2 are artificial because that tmTNF form contains mutations, which are not found in the native tmTNF molecule, and it is identical to solTNF. Also, tmTNF mutations to be retained on the cell membrane can differ, resulting in different outcome after an infection in vivo and in vitro [
30,
69]. At present, no data have been reported, excluding the interaction of TNFR1 with tmTNF. Many data have reported on TNFR1-tmTNF interactions in vivo and in vitro and, in particular, in the context of mycobacterial infections using tmTNF mutant mice that will be treated in this review.
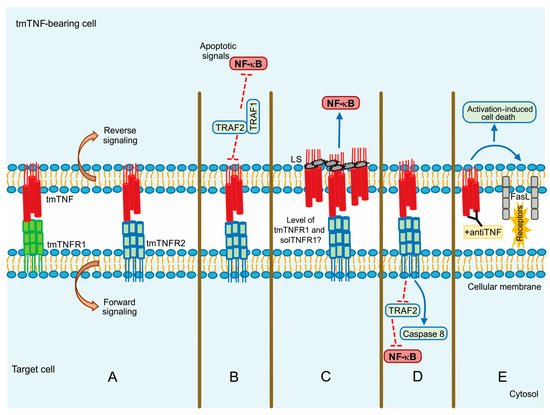
tmTNF is a molecule that induces crosstalk between tmTNF-bearing cells and tmTNFR-bearing cells signalling both as a ligand and as a receptor. This means that tmTNF not only mediates the forward signal to the target cell but also mediates the reverse signalling inside the tmTNF-bearing cell [
69] (A).
Figure 3. tmTNF signalling pathway. (A) Forward and reverse signalling; (B) Apoptotic signal by TRAF1 and TRAF2; (C) Direct NF-kB activation by LS (leader sequence) in tmTNF; (D) Actin involvement in tm-TNF transduction; (E) Activation Induced Cell Death (AICD) by upregulation of FASL (CD95L). Continuous blue lines: activation signalling. Dotted red lines: inhibition signalling.
Evidence suggests that although solTNF and tmTNF mediate cytotoxicity, tmTNF can exert apoptosis on solTNF-resistant cells. Using a model of TNF-resistant cells (the HL-60 cell line), it has been reported that tmTNF promotes the interaction with TRAF1 and TRAF2. TRAF1 plays a suppressor role in blocking the translocation of TRAF2 from the cytoplasm to the cell membrane. Consequently, NF-κB activation is inhibited resulting in cell death (B) [
70]. The same group has also proposed that forward signalling can result in opposite activities since tmTNF acting as a receptor promotes NF-κB activation, whereas acting as a ligand inhibits NF-κB activity [
71].
It has been reported that MCF-7 human tumor cells with a high expression level of tmTNF are resistant to cell death associated with solTNF and constitutive NF-κB activation. tmTNF contains a leader sequence (LS) in the cytoplasmic segment (76 amino acid residues) through which tmTNF is anchored into the membrane. LS appears to affect both forward and reverse signalling, and it seems that LS induces directly the constitutive activation of NF-κB [
72,
73] (C).
Using a pleural cell model, we have previously shown that tmTNF but not solTNF controls the expression of TNFR2 on myeloid cells expressing tmTNF, TNFR1 also contributes but in a minor way. Besides, the inflammatory process of BCG-induced pleurisy was downregulated mainly by tmTNF and TNFR2 [
74]. Recent studies have shown that tmTNF efficiently activates both TNFR1 and TNFR2, but solTNF interacts and activates TNFR1 [
75,
76]. In several liver injury models, it has been shown that only solTNF causes liver toxicity but not tmTNF which can protect the host against mycobacterial infections [
77,
78]. Recently, we reported that TNFR1 is necessary to recruit myeloid cells, while TNFR2 is implicated in cell activation after BCG-induced pleural infection [
78,
79].
The actin cytoskeleton plays an essential role in different functions of the cell, for instance, intracellular trafficking and cellular contractility, but also it has been recognized that the actin’s dynamic structure is involved in apoptosis and necrosis [
80]. The actin’s dynamic is another molecular mechanism by which the signalling activated can differ between both forms of TNF. Data have shown that solTNF induces actin depolymerization and morphological changes through ERK activation and p38 MAPK inducing cell death [
81]. In contrast, tmTNF does not affect the state of actin microfilaments; apparently, actin is involved in tmTNF-mediated signal transduction by uncoupling TRAF2 and cFLIP from TNFR2 and consequently activating caspase-8 to induce apoptosis and inhibiting NF-κB activation (D) [
82].
Activation-induced cell death (AICD) plays a role in regulating peripheral immune tolerance by deleting overactivated or autoreactive T cells. It has been described that NF-κB is required to mediate the expression of the pro-apoptotic molecule called Fas ligand (FasL or CD95L) inducing AICD [
83]. It was recently reported that when tmTNF functions as a receptor, using an anti-TNF polyclonal antibody to trigger reverse signalling, tmTNF can upregulate FasL expression and, consequently, increase AICD. Moreover, tmTNF-dependent reverse signalling also significantly increases several ligands, including TNFRs and Fas (E) [
84].