Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Medicine, Research & Experimental
It is now known that vascular endothelial growth factor (VEGF) and vascular endothelial growth factor receptors (VEGFRs) play a pivotal role in angiogenesis process [3-7]. Nowadays, the use of inhibitors of angiogenesis promoting factors is a powerful tool in anticancer combination therapeutic strategies, especially in cancer anti-angiogenic therapy (AAT).
- VEGF
- VEGF receptors
- anti-angiogenic therapy
1. VEGF Glycoproteins
The first reports on VEGF appeared in 1980s, when it was recognised as vascular permeability factor [48], vasculotropin [49] and, as currently known, vascular endothelial growth factor [50], an endogenous effector of prominent pro-angiogenic action through direct activation of vascular endothelial cells. VEGF belongs to the mammalian peptide family consisting of constituents originating from different genes: VEGF-A, VEGF-B, VEGF-C, VEGF-D and PlGF (placenta growth factor), but also viral homolog VEGF-E [51] and VEGF-F of snake venom origin [52]. The common feature of these glycoproteins is the creation of dimeric forms through specific sequence of cysteines forming disulphide bridges between two monomers [53]. Each VEGF family protein occurs as a glycosylated peptide monomer; however, it has to homodimerise or heterodimerise to activate its biological function.
VEGF-A (commonly called VEGF), is the most researched representative of the family and occurs in multiple isoforms (e.g., VEGF-A121, VEGF-A145, VEGF-A165, VEGF-A183, VEGF-A189 and VEGF-A206) due to an alternative splicing of mRNA obtained in the transcription process of the human gene VEGFA [54,55]. The VEGFA gene consists of eight exons that are highly conserved between species. In the first five constitutive exons are encoded the fundamental signal sequence, dimerisation cysteine fragment, specific VEGF receptors recognition domain, fragment employed in glycosylation and plasmin cleavage site, respectively. Furthermore, exons 6 and 7 encode an alternative heparine binding sequence and neuropilin binding domain, while last exon 8 encodes the unique VEGF domain. Alternative splicing results in variability of the primarily structure between isoforms, which affects their bioavailability and biological potency, mainly due to the isoform affinity to heparin sulphate and proteoglycan present on the extracellular surface competing with VEGF receptors [56]. Therefore, VEGF-A121 is freely diffusible and highly active isoform because it binds to neither neuropilins nor heparin sulphate, while VEGF-A165 and VEGF-A189 bind to both, resulting in expansion of their retention on the cellular surface or extracellular matrix.
Althought VEGF-A is highly recognised as a critical angiogenic inductor, it shows broad pleiotropic action in mammals, namely,
(I) significant mitogenic effect on vascular endothelial cells [57], as well as anti-apoptotic impact on these cells [58];
(II) increase of vascular permeability, resulting in increased serum peptides extravasation and local intra-tissue pressure [59];
(IV) neurotrophic and neuroprotective action [63].
The production of VEGF-A glycoproteins occurs in the endothelium and vascular smooth muscle cells, but also in activated platelets, fibroblasts, lymphocytes and macrophages [64], where the production may be stimulated by numerous factors. This process is especially noticeable in tumour cells, that hyperexpress VEGF to stimulate the promotion of tumour growth neoangiogenesis [65]. The main initiator of the transcription of mRNA encoding VEGF-A is hypoxia state, especially noticeable in the necrotic and cancer cells [66]. This phenomenon is associated with the formation of hypoxia induced factor in these cells, which is called hypoxia inducible factor-1 (HIF-1) [67,68]. In contrast to hypoxia, HIF-1 cellular concentration is strictly regulated under physiological conditions. Other significant stimulating factors of VEGF-A cellular synthesis are cytokines (interleukin 1b, IL-1b and tumour necrosis factor alpha, TNF-α), several hormones and specific growth factors [69,70], activation of oncogenes RAS and SRC, mutation in suppressor genes p53 and von Hippel–Lindau (VHL) [70,71,72], as well as nitric oxide and oxygen radicals [73,74]. These factors are more or less known as indirect initiators of angiogenesis, acting on the synthesis of VEGF-A.
The activity of other mammalian VEGF proteins is more specific than that of VEGF-A, however effects in site of action of all VEGF glycoproteins are more or less similar. VEGF-B has a relatively limited angiogenic action only towards ischemic myocardium, which is associated with VEGF-B level decrease [75]. More recently, it has been revealed that potent metabolic and antioxidative action of VEGF-B is possibly related to pro-angiogenic effects [76,77,78]. It contributes to the homeostasis of lipids in numerous tissues and the upregulation of brown adipose tissue, resulting in reduced risks of obesity and insulin resistance induced by diet rich in fat. Moreover, there are also reports of neuroprotective activity of exogenous VEGF-B186 isoform in the distal neuropathy and Parkinson’s disease models [79,80]. This effect is assumed to be induced directly on the motor neurons, similar to VEGF-A, not through their vascularity.
Some similarities to VEGF-B action exhibits placenta growth factor. PlGF is expressed dominantly by placental trophoblasts, but also during early embryonic development and to a lesser extent in a few adult organs such as heart, lungs, thyroid or skeletal muscles [81]. Contribution of PlGF in physiological angiogenesis in adults is negligible, however under pathological conditions such as ischemia, it prominently stimulates vascular endothelium proliferation and also differentiation and activation of the monocytes into the macrophages recognised as an angiogenic feedback stimulant [82]. Moreover, PlGF increases vessel permeability and inflammation in degenerations as rheumatoid arthritis and atherosclerosis promoting neoangiogenesis [83]. In addition, several types of tumour cell lines have the ability of PlGF expression, which favours the pro-angiogenic M2-phenotype tumour-associated macrophages [84].
VEGF-C is recognised as the fundamental promotor of proliferation and migration of the lymphatic system endothelium [85]. It also stimulates the cytokine-inducted migration and permeability of the vascular endothelial cells, although to a lesser extent than VEGF-A and independently of hypoxia stimulus. Similar in structure and function to VEGF-C, VEGF-D plays a secondary role in the physiological stimulation of human endothelium of vascular and lymphatic systems. Concomitantly, the high expression of both growth factors significantly promote and correlate with the metastasis through the lymphatic vessels in a variety of cancers [86,87,88].
2. VEGF Receptors and Their Co-Receptors
The site of action of VEGF glycoproteins are their specific receptors presented on the surface of target cells. There are three such receptors: VEGFR-1 (also known as FLT1, due to the same name of its gene), VEGFR-2 (known as KDR or FLK1, encoded by KDR gene) and VEGFR-3 (FLT4, encoded by FLT4 gene).
VEGFRs are classified as members of receptor tyrosine kinase superfamily due to their autophosphorylation ability induced by recognition of specific ligands. They are present in the form of homo- or heterodimers consisting of three functional fragments defined as extracellular part with seven Ig-like subunits, lipophilic single transmembrane domain and intracellular domain with distinctive tyrosine kinase activity. Individual VEGF proteins (and their isoforms) have different affinity towards each receptor. It is well known that VEGFR-1 binds VEGF-A, VEGF-B and PlGF, while VEGFR-2 binds VEGF-A as well as post-proteolytic VEGF-C and VEGF-D. Both VEGF-C and VEGF-D have affinity mainly towards VEGFR-3 [89] (Figure 1).
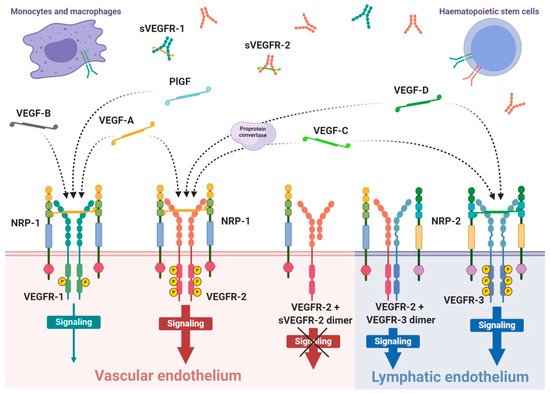
Figure 1. Scheme of expression of VEGF receptors and specificity of VEGF ligands. VEGF receptors occurs mainly as the homodimer transmembrane receptor tyrosine kinases, known as VEGFR-1, VEGFR-2 and VEGFR-3, or in soluble forms defined as sVEGFR-1 or sVEGFR-2. Moreover, surface receptors can create mixed heterodimers or even dimerise with soluble forms. VEGFR-1 expression occurs on vascular endothelium as well as haematopoietic stem cells, macrophages and monocytes. Expression on VEGFR-2 occurs mainly on vascular endothelium, less often on lymphatic endothelium, as well as on the surface of haematopoietic stem cells. The third receptor is mosty expressed on lymphatic endothelium. Conjugation of soluble form with transmembrane receptor preclude VEGF-driven signaling inside the cell. The mammalian VEGF glycoproteins, VEGF-A, VEGF-B, VEGF-C, VEGF-D and PlGF, are expressed as dimers that create different interations with specific VEGFRs, which is indicated by the dashed arrows. Representative VEGF-A glycoprotein binds to VEGFR-1 and VEGFR-2 with significantly higher affinity towards the first receptor. Concomitantly, VEGFR-1 is a specific molecular target for VEGF-B and PlGF, while VEGF-C and VEGF-D selectively bind to VEGFR-3; however, after proteolytic maturation, both VEGF-C and VEGF-D can also bind to VEGFR-2.
Interaction of growth factor with its receptor becomes much stronger with the participation of specific co-receptors that facilites the creation of the molecular complex ligand-receptor [90]. These co-receptors, known as neuropilins, occur as neuropilin 1 (NRP-1) that participates in VEGFR-1 or VEGFR-2 interactions with ligands and neuropilin 2 (NRP-2) mostly assigned to VEGFR-3 (Figure 1). Both types of neuropilins are expressed on endothelial cells and specific types of tumours [90,91]. NRP-1 binding differs between VEGF isoforms, so that VEGF-A165 and VEGF-A189 create stronger complexes with VEGFR-2 and NRP-1 than VEGF-A121, which is deprived of NRP-1 binding domain [90]. Nevertheless, direct interaction of VEGF-A121 with NRP-1 can regulate endothelial cell migration and sprouting independently of specific VEGF receptors [92].
The expression of VEGFR-1 occurs predominantly on endothelial cells of blood vessels, but also on monocytes and macrophages, placental trophoblasts as well as renal mesangial cells [93,94]. Similarly, VEGFR-2 occurs mostly on blood vessel endothelium, as well as platelets, haematopoietic and retinal stem cells. Both receptors are clearly expressed on cell surfaces of solid cancers and haematopoietic system neoplasms [95,96]. VEGFR-3 expression is specified only on endothelial cells of lymphatic system [97]. Therefore, a substantial share of VEGFR-1 and VEGFR-2 on vascular endothelium shows their significant contribution in angiogenesis, while VEGFR-3 and NRP-2 highly contribute in lymphangiogenesis [89,98].
For ligand binding receptors require at least the first three Ig-like domains, however, not all must participate in ligand binding. Simultaneously, if the ligand binds to neuropilin, then the third and fourth domains of the receptor will also attach to neuropilin. Moreover, besides ligand interaction, receptors also have to dimerise to be able to transduct signals intracellularly [99,100]. When both conditions are met, ligand can trigger the mutual autophosphorylation of the receptor intracellular tyrosine subunits and activation of specific signalling pathways inside the cell.
Different ligands can stimulate various biological effects through activated receptors, as well as activation of VEGFR-1 and VEGFR-2 by VEGF-A cause a different induction of intracellular signalling pathways [58,100]. Activation of VEGFR-2 leads to stimulation of the cell cycle, proliferation, migration, cell differentiation, angiogenesis, increased permeability of blood vessels but also inhibition of the apoptotic death and up-regulation of VEGF-A synthesis in endothelial cells [58,101]. On the contrary, VEGF-A can bind to VEGFR-1, activating its low-efficient tyrosine kinases, which has insignificant influence on endothelial cells [100,102]. Despite the high abundance of this receptor on endothelium, second receptor exerts even 10-fold higher density on endothelial cells [100,103]. Concomitantly, VEGF-A has about 10-fold lower affinity to VEGFR-2 compared to VEGFR-1. Hence, it is suspected that VEGFR-1 acts as concomitant decoy receptor and uptakes VEGF-A before it can bind to adjacent VEGFR-2, ergo VEGFR-1 plays an angiogenic-regulation role [82,103]. However, the same receptor interaction with PlGF promotes VEGF-A pool for endothelial angiogenic action through VEGFR-2 [82] and can regulate transphosphorylation of VEGFR-2 [104], thus amplifying angiogenesis through VEGFR-2. VEGFR-1 signalling can also regulate paracrine release in the vascular endothelial cells of other tissue endothelium growth factors inducing intestinal organogenesis and morphogenesis before vascular flow formation [105].
All VEGF isoforms that bind selectively to VEGFR-2 are capable to elicit receptor autophosphorylation, thus triggering the activation of numerous intracellular signalling pathways (Figure 2) [58,100,103,106,107]. Phosphorylated receptor subunits bind many adaptor molecules such as Shb (SH2 domain-containing adapter protein B), SOS (Son of sevenless proteins) or Grb-2 (Growth factor receptor-bound protein 2) that activate Ras GPTase. This last protein stimulates MAPK pathway responsible for endothelium proliferation. Simultaneously, phosphorylated intracellular VEGFR-2 domain activates phospholipase C-gamma (PLC-γ), which catalyses hydrolysis of phosphatidylinositol bisphosphate (PIP2) to inositol triphosphate (IP3) and diacylglycerol (DAG). IP3 triggers intracellular release of Ca2+ form endoplasmic reticulum, which employs calcium modulated protein calmodulin to stimulate cAMP phosphodiesterase, adenylate cyclase and site-specific endothelial NO synthase (eNOS) and consequently increase NO-driven vasodilation and vascular permeability. However, DAG activates calcium-dependent protein kinase C (PKC), a multi-target kinase stimulating indirect cell proliferation and migration. Additionally, phosphorylated VEGFR-2 induces protein kinase B (commonly known as AKT) at the beginning of PI3K/AKT/mTOR pathway, an important signalling regulator of the cell cycle and metabolism, reducing risk of apoptosis and promoting cellular transcription, proliferation and migration [58,106]. Moreover, phosphorylated VEGFR-2 activates signalling of focal adhesion kinase (FAK) observed during cellular migration, adhesion, cytoskeleton rearrangement and tumour progression [107,108]. Nevertheless, it was observed that VEGF-A can regulate endothelial cell attachment independently of VEGFR-2 through NPR-1 [109].
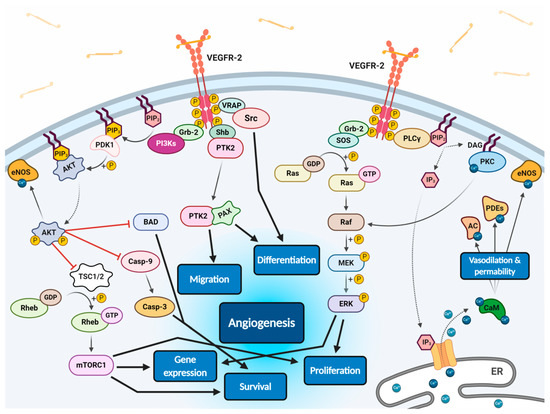
Figure 2. Scheme of endothelial signal transduction of VEGF-VEGFR-2 ligand-receptor molecular complex. The autophosphorylation of receptor tyrosine kinase domains caused by VEGF binding stimulates multiple specific VEGFR-associated proteins (VRAPs) and adaptor molecules inducing concurrent intracellular signalling pathways that promotes proliferation, differentiation, migration, gene expression and apoptosis survival of endothelium leading to angiogenesis.
VEGF receptors, in addition to transmembrane forms, can also occur in soluble forms, known as sVEGFR-1 and sVEGFR-2 (Figure 1) [108,110]. Their formation can be explained by two mechanisms, namely, a proteolysis of extracellular binding domain [111,112] and alternative splicing of primary gene transcript [108,113], both forming freely diffusible proteins consisting of only six of seven Ig-like subunits [114]. Soluble receptors are secreted by identical cells that express regular receptors, mostly by vascular endothelial cells [110]. Due to the fact that sVEGFRs exhibit comparable binding affinity on a similar basis as regular receptors, but are deprived of effector domains of tyrosine kinases, they can demonstrate only a regulatory decoy function. Both soluble receptors compete for VEGF-A with regular receptors inhibiting angiogenic and other actions of the growth factor. Simultaneously, sVEGFR-2 can uptake VEGF-C and VEGF-D reducing their overall supply intended for lymphangiogenesis stimulation through VEGFR-3 [113]. Moreover, creation of heterodimers from soluble and regular receptors precludes cellular signalling [110]; however, it is suspected that interaction of sVEGFRs with NRP-1 can mediate VEGF-A trigger of intracellular PKC pathway signalling [115].
Interestingly, several reports have shown the reverse correlation between sVEGFR expression and cancerous angiogenesis or metastasis. Such research has indicated that sVEGFR-1 permanently suppresses tumour growth and decreases metastasis promoting overall survival rate in rodents or humans with fibrosarcoma and glioblastoma [116], advanced renal cancer [117], breast cancer [118,119], acute myeloid leukaemia [120], colorectal cancer [121] and non-small cell lung cancer [122]. Similar results were presented for sVEGFR-2 [119,123,124,125,126], demonstrating significant biomarker role of these receptors in diagnosis of numerous cancers.
3. Anti-Angiogenic Therapy Strategies for Tumour Treatment
Although various angiogenesis-stimulating factors exist, VEGF-A is considered the most potent and predominant one. This also applies to sustained angiogenesis in cancers. Currently, it is known that angiogenesis, besides its crucial role in the tumour growth, stimulates the progression of invasiveness and development of vascular network in the surrounding tumour microenvironment [127,128]. The concept of angiogenesis targeting for cancer diagnosis and treatment seems promising, therefore, a wide variety of therapeutic strategies have been directed at visualisation and interfering with tumour-stimulated angiogenesis. However, since the first FDA approval of bevacizumab (BV), humanised anti-VEGF-A mAb, for the combinational chemotherapy regimen with 5-fluorouracil of metastatic colorectal cancer [129], only a few AAT strategies have been granted similar approval. It has become a challenge to evaluate these strategies almost personally for each patient, due to considerable variability of the angiogenic process in each treated entity [42]. Although the correlation between tumour progression and VEGF-A expression is well established, it does not transfer into intended anti-angiogenic therapeutic effects. This is due to the heterogeneity of the same tumour between patients, but also between different tumours in an individual patient, that occurs and changes at different stages of the lesion development. This raises the need for appropriate methods of assessing how the patient responds to the proposed therapy. In terms of AAT, this applies to clinically significant parameters as the lesion location with regard to tumour admission of therapeutic agents and expression of endogenous growth factors in tumour microenvironment affecting the saturation of target receptors involved in angiogenesis. Despite the complexity of this issue, the use of radiopharmaceuticals is increasingly proposed for independent preliminary screening, which can provide the prediction of patient clinical response [130]. Radioligands successfully targeting VEGF/VEGFR system in vivo are potentially valuable tracers for the study of angiogenic processes [131], stratification of patients to AATs [132], as well as monitoring therapy efficacy and clinical outcomes [133,134].
Basically, the aforementioned radiopharmaceuticals are based on various approaches to VEGF/VEGFR system targeting including radiolabelled derivatives of human VEGF-A ligands, anti-VEGF or anti-VEGFR antibodies, VEGFR binding peptides, small molecular inhibitors of tyrosine kinase domain of VEGF receptors and peptidomimetic ligands targeting NRP-1 co-receptor. Additionally, depending on specific radiation features of applied radionuclide, the radiopharmaceuticals are dedicated for diagnostic, therapeutic or theranostic purposes. This multitude of radiopharmaceutical solutions allows for the design of tailor-made therapeutic tool and its evaluation on a specific cancer model. The broad selection of above listed biovectors enables choice of one that provides the desired multiple molecular targets or just specific one, exhibits eligible pharmacokinetics, predicts response of certain chemotherapeutic strategy, or shows confirmed complemental contribution to the selected chemotherapy.
AAT methods have especially found a place in clinical practice applied in monotherapy. Currently, it is well known that even these methods used alone are inefficient, they advantageously support conventional chemotherapy effects [135]. Interestingly, the AAT contributes to normalisation of the tumour vasculature resulting in enhanced metabolic rate and delivery capacity of the tumour; hence, AAT can increase efficacy of the radiotherapy or activity of immune system in the close tumour surroundings.
Funding
This research was carried out within grant 2019/33/B/NZ7/02818, supported by the National Science Centre (Poland).
Acknowledgments
The contributions of Katarzyna Masłowska and Paweł Krzysztof Halik have been done in the frame of the National Centre for Research and Development Project No POWR.03.02.00-00-I009/17 (Radiopharmaceuticals for molecularly targeted diagnosis and therapy, RadFarm, Operational Project Knowledge Education Development 2014–2020 co-financed by European Social Fund).
This entry is adapted from the peer-reviewed paper 10.3390/cancers13051072
This entry is offline, you can click here to edit this entry!