Limbic encephalitis (LE) is a rare cause of encephalitis presenting as an acute and subacute onset of neuropsychiatric manifestations, particularly with memory deficits and confusion as core features, along with seizure occurrence, movement disorders, or autonomic dysfunctions.
- limbic encephalitis
- neuropsychiatric
- LGI1
- GAD
- GABA
- autoimmune
1. Introduction
Limbic encephalitis (LE) is an inflammatory encephalitis involving the limbic system, which encompasses the medial temporal lobe, hippocampus, and frontobasal and cingulate cortex [1]. LE is frequently associated with antibodies against the neuronal cell surface, synaptic vesicles, and intracellular proteins; therefore, it belongs to the autoimmune encephalitis (AE) category. Triggering factors include tumors, viral infections, or immune check point inhibitors (ICI’s) [2,3]. LE most often occurs in adults older than 45 years but can affect people of all ages. In addition, gender predominance varies with the type of antibody.
The clinical hallmark of LE includes acute and subacute onset of memory and cognitive deficits. Other symptoms include confusion, psychiatric symptoms (such as anxiety, depression, or psychosis), behavioral changes, seizures, movement disorders (such as ataxia, dystonia, or myoclonus), autonomic disturbances, and sleep disturbances [1,4]. The amygdala is a core region of the limbic system involved in the control of positive and negative affect, modulation of memory, and social and cognitive functions as well as behavioral adaptation to stress [5], which explains the core neuropsychiatric manifestations of LE. The basolateral complex of the amygdala is the main input site for sensory information from the thalamus and cortical regions, which plays a central role in seizure generation, particularly in the temporal lobe epilepsy [6].
2. Antibodies
Antibodies associated with LE are thought to be of peripheral origin that penetrates a leaky blood–brain barrier (BBB) or to be synthesized intrathecally. Most AE has higher serum than CSF antibody levels, implicating that antibodies are likely to be initiated by a peripheral immune response [12]. The autoantibodies are predominantly of IgG1 subclass. However, LGI1 and CASPR2 antibodies are mainly IgG4 subclass [13,14]. IgG4 antibody is hetero-bispecific (continuously undergoing half antibody exchange), hence less effective than IgG1 in crosslinking and internalizing the target antigen and does not fix complement [15]. In general, IgG1-associated AE (e.g., N-methyl-D-asparate receptor [NMDAR], γ-aminobutyric acid-B receptor [GABA-BR], and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor [AMPAR]) have more inflammatory signs than IgG4-associated AE (e.g., LGI1 and CASPR2) [16]. Nevertheless, complement-mediated neuronal loss was still observed in LGI1 and CASPR2-associated neurologic syndromes [17,18]. These patients had higher complement-fixing IgG1 subclass with more prolonged clinical course, more severe cognitive impairment, and frequent hippocampal atrophy in advanced stages of the disease [13,19].
Antibodies associated with LE target: (1) cell-surface receptors (GABA, AMPA, and glycine receptors); (2) ion channels (voltage-gated potassium channels [VGKC]); (3) neighboring proteins that stabilize channel complex into the membrane (LGI1 and CASPR2); (4) enzymes that catalyze the formation of neurotransmitters glutamic acid decarboxylase (GAD); and (5) intracellular proteins (Hu, Ma2, collapsin response-mediator protein-5 [CRMP5], and amphiphysin) [20]. These antibodies are classified as neuronal cell surface, synaptic vesicle, and intracellular antibodies according to the location of their targeting antigens. Antibodies against intracellular antigens are also known as onconeural antibodies because of their large association with tumors, and cause paraneoplastic syndromes (PNS) [21]. Pathologic effects include direct blockage of receptors and ion channels, indirect disruption with neighboring molecules interaction, and crosslinking and internalization of receptors to deplete them from the cell surface (Figure 1).
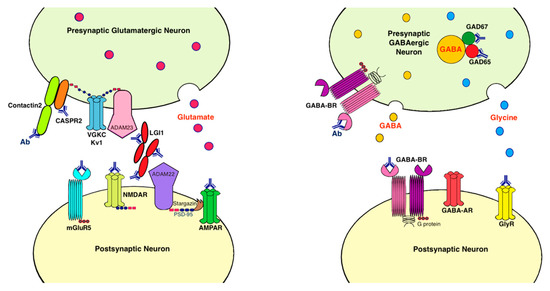
Figure 1. Antibodies and their pathogenic effects in limbic encephalitis. The pathologic effects of antibodies include blocking of receptors or ion channels, disruption of interaction with neighboring molecules, and crosslinking and internalization of receptors from cell surface. (1) CASPR2 and contactin2 antibodies inhibit the interaction between these proteins and reduce the clustering and surface expression of VGKC; (2) LGI1 antibody disrupts interaction between protein components such as LGI1 to ADAM22/23, downregulates VGKC and reduces AMPAR clustering and synaptic transmission; (3) AMPAR antibody causes cross-linking and receptor internalization; (4) NMDAR* antibody causes cross-linking and receptor internalization; (5) mGluR5 antibody causes decreased synaptic mGluR5 cluster density; (6) GAD antibodies target mostly the GAD65 subunits but also the GAD67 subunits, disrupting GABAergic signaling; (7) GABA-BR antibody prevents ligand binding to the receptor and alters receptor function; (8) GlyR antibody probably acts as antagonist to disrupt receptor function. Ab: antibody; ADAM: a disintegrin and metalloproteinase; AMPAR: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor; CASPR2: contactin associated protein-like 2; GABA-AR: γ-aminobutyric acid-A receptor; GABA-BR: γ-aminobutyric acid-B receptor, GAD: glutamic acid decarboxylase; GlyR: glycine receptor; LGI1: leucine-rich, glioma inactivated 1; mGluR5: metabotropic glutamate receptor 5; NMDAR: N-methyl-D-asparate receptor; PSD-95: postsynaptic density protein 95; VGKC: voltage-gated potassium channels.
3. Immunopathology
4. Inflammatory Mediators
Conventional CSF inflammatory markers such as white blood cells or protein are neither sensitive nor specific for LE, although the detection of oligoclonal band may increase diagnostic sensitivity [44]. A lower CD4/CD8+ T-cell ratio is detected in the CSF of all patients with LE [45].
The serum and CSF levels of C-X-C motif chemokine ligand 13 (CXCL13) were significantly higher in patients with LGI1 encephalitis [46]. The serum levels of CXCL10 were elevated in CASPR2 encephalitis. CXCL10 is a cytokine that recruits C-X-C motif chemokine receptor 3 cells such as activated T cells [47]. CASPR2 encephalitis seems to elicit a higher immune response than LGI1 encephalitis, with more intrathecal IgG and higher cytokine levels, indicated by higher CXCL13 and soluble intercellular adhesion molecule-1 (sICAM1) in the CSF. CXCL13 points to a B-cell mediated, whereas sICAM1 to a T-cell mediated neuroinflammation [48]. Patients with PNS also had high levels of chemokine CXCL10 in the CSF with the presence of IFN-γ receptors on their T cells [47].
CSF mediators are different in infectious and immune-mediated encephalitis [49]. CSF cytokines IL-21 and IFN-γ-induced protein 10 kDa (IP10) are promising in differentiating between viral encephalitis and AE [50]. IL-21 causes autoantibody production, which downregulates regulatory T cells leading to enhanced autoimmunity and increased CD8+ T cells and NK cells in AE [51], whereas IP10/CXCL10 is secreted in response to IFN-γ, which is produced as part of the Th1 in response to viral infection [52].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22010389