Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Chemistry, Medicinal
Veratrum californicum contains steroidal alkaloids that function as inhibitors of hedgehog (Hh) signaling, a pathway involved in the growth and differentiation of cells and normal tissue development. This same Hh pathway is abnormally active for cell proliferation in more than 20 types of cancer.
- Veratrum californicum
- alkaloid
- hedgehog signaling pathway
- bioactivity
- cyclopamine
1. Introduction
Few treatments exist outside of surgery for basal cell carcinomas (BCCs), and the drug therapies which are available are considered a last resort [1][2]. Patients often discontinue drug therapy prematurely due to extreme side effects including alopecia, fatigue, muscle spasms, and others [1][2]. An under-explored source of potential treatment exists in the high mountain meadows of Idaho. Veratrum californicum contains steroidal alkaloids that are potent hedgehog (Hh) signaling inhibitors [3]. The Hh signaling pathway is implicated in the growth of at least 20 types of cancer, and drug treatment has recently focused on targeting this pathway [4]. Cyclopamine, the most well-studied alkaloid extracted from V. californicum, has been used to understand the mechanism of the Hh signaling pathway in cancer progression and has served as a molecular scaffold for modern chemotherapeutics [2][5][6][7]. Early studies of V. californicum only examined major components of the root and rhizome raw extract [8][9][10][11][12]. The focus of this current study is to gain insight into the less abundant alkaloids in the V. californicum root and rhizome in order to discover alkaloids that suppress Hh signaling.
2. Veratrum Californicum Alkaloids
During the 1950s, in the mountain meadows of Idaho, sheepherders reported that up to a quarter of their newborn sheep had a craniofacial malformation [5][13]. Deformities affecting their skulls, jaws, sometimes brains, and eyes led to the term “monkey-faced” lambs [13]. The most characteristic feature was a singular, enlarged, cyclopean eye in the middle of the face [13]. In 1954 the Poisonous Plant Research Laboratory (PPRL) in Logan, Utah, was given the task of discovering the origin of these mutations [5]. At the time the cause of the abnormalities was proposed to be a recessive genetic trait, but a breeding study in 1957 eliminated this possibility [13]. The next step in determining the source was performing field and feeding studies [5].
V. californicum was explored as a potential cause of the lambs’ alterations in 1958 after a sheepherder observed it causing the sheep to become sick following consumption. Feeding trials began soon after, and the PPRL reported the sheep experiencing a variety of infirmities, including excessive salivation and frothing at the mouth, vomiting, abnormal gate, irregular heartbeat, dyspnea, convulsions, coma, death, and in 1959 a cyclopean eye was produced. After several more years of trials, a definitive correlation was made between the ewes’ consumption of V. californicum on day 14 of gestation to lambs born with cyclopean malformations underneath a proboscis-like nose [3][5].
In the search for the molecular explanation of these symptoms, the alkaloid cyclopamine was separated from V. californicum raw extract in 1968, and in 1969 its steroidal structure was published [10][11]. Cyclopamine was identified by Japanese scientists in 1965 from the extract of Veratrum grandiflorum, and it had been given the name 11-deoxojervine [5]. It was concluded that the three alkaloids cyclopamine, cycloposine, and jervine were responsible for the lambs’ deformities. Previously, other Veratrum alkaloids (cevanine-type) had been studied for their hypotensive properties, but it became clear by 1970 that some of these newer discoveries were teratogenic [3]. V. californicum alkaloid discovery continued for a few more years, and then remained relatively untouched until the early 2000s.
Eventually more precise instrumentation and techniques became available for solid phase extraction, which greatly improved the extraction, separation, isolation, characterization, and bioactivity assessment of V. californicum alkaloids [14]. Over the past decade, the lab has explored less abundant alkaloids from V. californicum that were under-studied [15]. This work began with a comparison of eight methods for extracting cyclopamine from V. californicum, which led to the conclusion that ethanol extraction produced the highest number of alkaloids, with the greatest retained bioactivity [16]. Next, cyclopamine, veratramine, muldamine, and isorubijervine were quantified in the different plant parts: roots/rhizomes, stems, and leaves [17]. Additionally, the variation in alkaloid content was explored for the various plant parts, stages of growth, and harvest locations. This led to the detection of six new uncharacterized alkaloids, all of which appeared to exhibit Hh pathway suppression activity, thus inspiring the current study [4].
3. Hedgehog Signaling Pathway
V. californicum alkaloids are of interest due to their inhibition of the Hh signaling pathway, which is critical for intercellular communication during fetal development [18][19][20]. The Hh pathway involves proper cell polarization, most epithelial tissue differentiation leading to bilateral symmetry, and the formation of the central nervous system, skeleton, limbs, teeth, eyes, and several vital organs within vertebrate embryos [4][5][19][21][22][23][24]. Loss of the pathway’s proper function can result in a range of complications from under-developed facial features to cyclopia to nervous system disorders [5]. In addition, propagation of over 20 cancers has been correlated to aberrant Hh signaling. Blocking the Hh signaling pathway during development can be detrimental, but inhibition in cancer is a treatment option [3][4].
The Hh gene was first discovered in 1978 through Drosophila gene knockout trials conducted by Christiane Nusslein-Volhard and Eric Wieschaus, who later earned a Nobel Prize for their work. When the Hh gene was silenced, the fruit fly embryo was short with spine-like projections reminiscent of a hedgehog [5][25]. The Hh pathway in vertebrates utilizes three signaling molecules, which were named after hedgehog breeds and a fictional character: Desert hedgehog (Dhh), Indian hedgehog (Ihh), and Sonic hedgehog (Shh) [5]. These molecules must be secreted with the help of the membrane protein Dispatched (DISP1), cholesterol, and secreted protein SCUBE2 (Figure 1, 1.) [26][27]. When Hh protein binds Patched 1 (PTCH)—a 12-span transmembrane protein—inhibition of Smoothened (SMO)—a seven-span transmembrane protein—is interrupted, and a signaling cascade is initiated (Figure 1, 2.) [5][18][26][27][28][29][30][31][32]. SMO is phosphorylated using GRK2 and casein kinase 1 (CK1) and begins to collect in the primary cilium (Figure 1, 3.) [26][28]. This causes the inhibition of Suppressor of Fused (SuFu) and Kif7, which then release a glioma associated oncogene homolog (Gli) (Figure 1, 4.) [26][28][29][30]. The levels of zinc-finger transcription factors from the cubitus interruptus (Ci)/Gli family determine if the Hh signal is transmitted [5][18]. Gli 1 and Gli 2 function as transcriptional activators (Figure 1, 5.), while Gli 3 is a repressor [5][18][26][28][29][30][31]. When the pathway is off, PTCH inhibits SMO, allowing SuFu and Kif7 to sequester Gli (Figure 1, 6. and 7.) [26][28][29][30][31]. SMO is marked for degradation, and SuFu and Gli are phosphorylated using protein kinase A (PKA), glycogen synthase kinase 3 (GSK3), or CK1 (Figure 1, 8. and 9.) [26][29][30][32]. Gli can then take one of two paths, which are mediated using β-TRCP, an E3 ubiquitin ligase, and its adaptor protein, SPOP [28]. First, it can be marked for degradation and passed onto the proteosome, and second, it can be proteolytically cleaved and enter the nucleus as Gli3, inhibiting Hh gene transcription (Figure 1, 10. and 11.) [30][31]. Another method of inhibiting transcription is by using the teratogenic alkaloid cyclopamine, the first discovered Hh pathway suppressor, which replaces PTCH, stopping signal transduction [5][18].
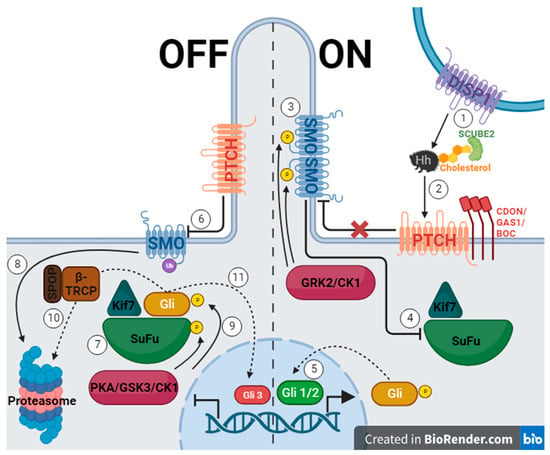
Figure 1. The hedgehog signaling pathway, where the left side of the figure represents the off state of the pathway due to PTCH inhibiting SMO, and the right side represents the on state when the pathway is activated by Hh protein binding to PTCH allowing SMO to function. (Created with BioRender.com).
Cyclopamine has been used to inhibit the Hh signaling pathway when it is irregularly activated by cancer [3][5][33]. In mice, cyclopamine has suppressed cancerous tumor growth in several models, including xenografts models of human colon cancer [34], glioma [35], melanoma [36], pancreatic cancer [37][38][39], prostate cancer [40], and small cell lung cancer [41], in addition to a medulloblastoma allograft [42], and a genetic medulloblastoma model [5][43]. In humans, cyclopamine has been infused into an oil-based ointment for topical application directly to tumors, which has led to tumor size reduction without detrimental side effects [5].
As a drug therapy, cyclopamine does have some weaknesses: it is not well solubilized in water or physiological environments, it degrades in acidic environments (e.g., stomach acid), and it can inhibit cellular neurogenesis and proliferation as adult neural stem cells still utilize the Hh pathway into maturity [4][5][33][44][45]. Exploration for new Hh inhibitor therapies began with the structure of cyclopamine as a prototype. This work started at Johns Hopkins University, where a 3-keto N-(aminoethyl-aminocaproyl-dihydrocinnamoyl) moiety was combined with cyclopamine (KAAD-cyclopamine) to increase its solubility, and in mouse models the new molecule was 10–20 times more potent than cyclopamine alone [5]. Further studies based on cyclopamine’s molecular scaffold were conducted to see if the side effects could be reduced, which resulted in IPI-926 (a.k.a. patidegib), a semi-synthetic variant with increased stability and potency [7]. Phase 3 clinical trials have been completed for IPI-926 in a 2% topical gel for the treatment of basal cell carcinomas for patients with basal cell nevus syndrome (Gorlin syndrome), but the results were not reported [46]. Computational research modeled off cyclopamine was performed to identify additional molecules of interest. In one instance, a multitude of small molecules were screened for their ability to bind SMO [6]. In 2012, FDA approval was given for Vismodegib (GDC-0449 or Erivedge) to treat metastatic basal cell carcinoma (mBCC) or locally advanced basal cell carcinoma (laBCC) in adults unable to undergo surgery or radiation therapy [6][47]. Developed by Genentech, Inc., Vismodegib had an overall response rate (ORR) of 60% in patients with laBCC and 46% in patients with mBCC in Phase II clinical trials. In this study, 30% of mBCC patients demonstrated a decrease in tumor size, and 43% of laBCC patients displayed lesions healing or a significant decrease in tumor size. Common side effects included decreased appetite, diarrhea, dysgeusia, fatigue, hair loss, muscle spasms, and nausea [6]. A small-molecule in vitro screening performed by Sun Pharma Global resulted in Sonidegib (LDE225, erismodegib, or ODOMZO) receiving FDA approval in 2015 to treat recurrent advanced basal cell carcinoma (aBCC) in patients unable to undergo surgery or radiation [2][33][48]. In Phase II clinical trials, after 12 months of treatment, patients with laBCC (18 of 94) either died or had their condition progress, but the ORR was 57.6% with 200 mg and 43.8% with 800 mg. For patients with mBCC the ORR was 7.7% for 200 mg and 17.4% for 800 mg [49]. From most to least common, the potential side effects of taking Sonidegib were muscle spasms, alopecia, dysgeusia, nausea, increased creatinine kinase, fatigue, weight loss, diarrhea, decreased appetite, myalgia, and vomiting [2]. Eventually, patients experienced resistance to both Vismodegib and Sonidegib as a result of a SMO mutation or the initiation of an alternative Hh pathway [50]. In 2018, Pfizer’s Glasdegib (PF-0449913 or DAURISMO™) in combination with low-dose cytarabine (LDAC) was approved by the FDA as an oral treatment for acute myeloid leukemia (AML) patients of at least 75 years of age or patients with co-morbidities who cannot undergo intensive induction chemotherapy [33][51]. In the BRIGHT AML 1003 study, when contrasted with only LDAC, Glasdegib paired with LDAC demonstrated a decrease in mortality by 54%, but patients experienced pneumonia, fatigue, dyspnea, hyponatremia, sepsis, and syncope [51]. The molecular structures for each of the alkaloids (cyclopamine, KAAD-cyclopamine, and IPI-926) and chemotherapeutics (Vismodegib, Sonidegib, and Glasdegib) are shown in Figure 2.
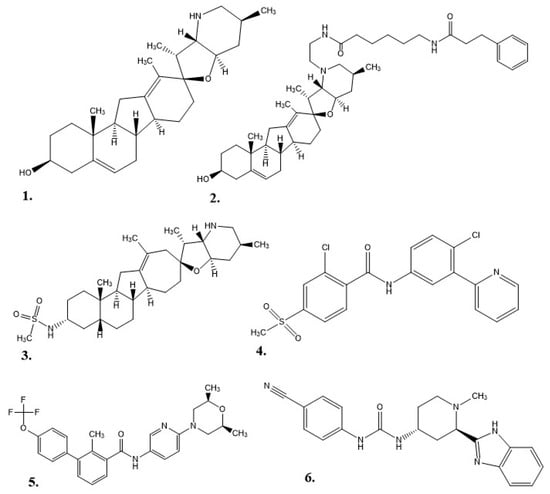
Figure 2. Structures of hedgehog signaling pathway inhibitors. (1) Cyclopamine, (2) KAAD-cyclopamine, (3) IPI-926, (4) Vismodegib, (5) Sonidegib, (6) Glasdegib.
With cyclopamine being the first identified molecule with the ability to block Hh signaling and cyclopamine variants exhibiting increased Hh signaling suppression, it is reasonable to revisit V. californicum in the quest for more inhibitors that may have been overlooked due to their low natural abundance at the time of the original work in the 1950s. Preliminary work in the lab resulted in the detection of sixteen alkaloids, where six were identified using commercially available standards, and the identities of five others were speculated based on the predicted molecular formula obtained using mass spectrometry (MS) from the mass to charge (m/z) ratio [4][17]. The bioactivity work demonstrated that the raw root/rhizome extract was more effective at Hh signaling inhibition than a proportionate amount of cyclopamine [4][52].
This entry is adapted from the peer-reviewed paper 10.3390/ph17010123
References
- Lauressergues, E.; Heusler, P.; Lestienne, F.; Troulier, D.; Rauly-Lestienne, I.; Lie Tourette, A.; Ailhaud, M.-C.; Cathala, C.; Phanie Tardif, S.; Denais-Laliè Ve, D.; et al. Pharmacological Evaluation of a Series of Smoothened Antagonists in Signaling Pathways and after Topical Application in a Depilated Mouse Model. Pharmacol. Res. Perspect. 2016, 4, 214.
- Jain, S.; Song, R.; Xie, J. Sonidegib: Mechanism of Action, Pharmacology, and Clinical Utility for Advanced Basal Cell Carcinomas. OncoTargets Ther. 2017, 10, 1645–1653.
- Chandler, C.M.; McDougal, O.M. Medicinal History of North American Veratrum. Phytochem. Rev. 2013, 13, 671–694.
- Turner, M.W.; Rossi, M.; Campfield, V.; French, J.; Hunt, E.; Wade, E.; McDougal, O.M. Steroidal Alkaloid Variation in Veratrum Californicum as Determined by Modern Methods of Analytical Analysis. Fitoterapia 2019, 137, 104281.
- Lee, S.T.; Welch, K.D.; Panter, K.E.; Gardner, D.R.; Garrossian, M.; Chang, C.W.T. Cyclopamine: From Cyclops Lambs to Cancer Treatment. J. Agric. Food Chem. 2014, 62, 7355–7362.
- Aditya, S.; Rattan, A. Vismodegib: A Smoothened Inhibitor for the Treatment of Advanced Basal Cell Carcinoma. Indian. Dermatol. Online J. 2013, 4, 365.
- Tremblay, M.R.; Lescarbeau, A.; Grogan, M.J.; Tan, E.; Lin, G.; Austad, B.C.; Yu, L.C.; Behnke, M.L.; Nair, S.J.; Hagel, M.; et al. Discovery of a Potent and Orally Active Hedgehog Pathway Antagonist (IPI-926). J. Med. Chem. 2009, 52, 4400–4418.
- Keeler, R.F.; Binns, W. Chemical Compounds of Veratrum Californicum Related to Congenital Ovine Cyclopian Malformations: Extraction of Active Material. Proc. Soc. Exp. Biol. Med. 1964, 116, 123–127.
- Keeler, R.F.; Binns, W. Teratogenic Compounds of Veratrum Californicum (Durand). I. Preparation and Characterization of Fractions and Alkaloids for Biologic Testing. Can. J. Biochem. 1966, 44, 819–828.
- Keeler, R.F. Teratogenic Compounds of Veratrum Californicum (Durand)—IV. Phytochemistry 1968, 7, 303–306.
- Keeler, R.F. Teratogenic Compounds of Veratrum Californicum (Durand)—VI. The Structure of Cyclopamine. Phytochemistry 1969, 8, 223–225.
- Keeler, R.F. Teratogenic Compounds of Veratrum Californicum (Durand) VII. The Structure of the Glycosidic Alkaloid Cycloposine. Steroids 1969, 13, 579–588.
- Binns, W.; James, L.F.; Shupe, J.L.; Thacker, E.J. Cyclopian-Type Malformation in Lambs. Am. Med. Assoc. 1962, 5, 106–108.
- Oatis, J.E.; Brunsfeld, P.; Rushing, J.W.; Moeller, P.D.; Bearden, D.W.; Gallien, T.N.; Cooper IV, G. Isolation, Purification, and Full NMR Assignments of Cyclopamine from Veratrum Californicum. Chem. Cent. J. 2008, 2, 12.
- Chandler, C.M.; Habig, J.W.; Fisher, A.A.; Ambrose, K.V.; Jiménez, S.T.; Mcdougal, O.M. Improved Extraction and Complete Mass Spectral Characterization of Steroidal Alkaloids from Veratrum Californicum. Nat. Prod. Commun. 2013, 8, 1059–1064.
- Turner, M.W.; Cruz, R.; Mattos, J.; Baughman, N.; Elwell, J.; Fothergill, J.; Nielsen, A.; Brookhouse, J.; Bartlett, A.; Malek, P.; et al. Cyclopamine Bioactivity by Extraction Method from Veratrum Californicum. Bioorganic Med. Chem. 2016, 24, 3752–3757.
- Turner, M.W.; Cruz, R.; Elwell, J.; French, J.; Mattos, J.; McDougal, O.M. Native V. Californicum Alkaloid Combinations Induce Differential Inhibition of Sonic Hedgehog Signaling. Molecules 2018, 23, 2222.
- Gupta, S.; Takebe, N.; LoRusso, P. Targeting the Hedgehog Pathway in Cancer. Ther. Adv. Med. Oncol. 2010, 2, 237.
- Carballo, G.B.; Honorato, J.R.; de Lopes, G.P.F.; de Sampaio e Spohr, T.C.L. A Highlight on Sonic Hedgehog Pathway. Cell Commun. Signal. 2018, 16, 11.
- Cooper, M.K.; Porter, J.A.; Young, K.E.; Beachy, P.A. Teratogen-Mediated Inhibition of Target Tissue Response to Shh Signaling. Science 1998, 280, 1603–1607.
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and Mechanisms. Genes Dev. 2008, 22, 2454–2472.
- Echelard, Y.; Epstein, D.J.; St-Jacques, B.; Shen, L.; Mohler, J.; McMahon, J.A.; McMahon, A.P. Sonic Hedgehog, a Member of a Family of Putative Signaling Molecules, Is Implicated in the Regulation of CNS Polarity. Cell 1993, 75, 1417–1430.
- Krauss, S.; Concordet, J.P.; Ingham, P.W. A Functionally Conserved Homolog of the Drosophila Segment Polarity Gene Hh Is Expressed in Tissues with Polarizing Activity in Zebrafish Embryos. Cell 1993, 75, 1431–1444.
- Riddle, R.D.; Johnson, R.L.; Laufer, E.; Tabin, C. Sonic Hedgehog Mediates the Polarizing Activity of the ZPA. Cell 1993, 75, 1401–1416.
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations Affecting Segment Number and Polarity in Drosophila. Nature 1980, 287, 795–801.
- Cell Signaling Technology. Hedgehog Signaling. Available online: https://www.cellsignal.com/pathways/hedgehog-signaling-pathway (accessed on 26 July 2023).
- Tukachinsky, H.; Kuzmickas, R.P.; Jao, C.Y.; Liu, J.; Salic, A. Dispatched and Scube Mediate the Efficient Secretion of the Cholesterol-Modified Hedgehog Ligand. Cell Rep. 2012, 2, 308–320.
- Abcam. Hedgehog Signaling Pathway: An Overview. Available online: https://www.abcam.com/pathways/hedgehog-signaling-pathway (accessed on 26 July 2023).
- Bhateja, P.; Cherian, M.; Majumder, S.; Ramaswamy, B. The Hedgehog Signaling Pathway: A Viable Target in Breast Cancer? Cancers 2019, 11, 1126.
- Yang, C.; Qi, Y.; Sun, Z. The Role of Sonic Hedgehog Pathway in the Development of the Central Nervous System and Aging-Related Neurodegenerative Diseases. Front. Mol. Biosci. 2021, 8, 711710.
- Jeng, K.S.; Jeng, C.J.; Jeng, W.J.; Sheen, I.S.; Li, S.Y.; Leu, C.M.; Tsay, Y.G.; Chang, C.F. Sonic Hedgehog Signaling Pathway as a Potential Target to Inhibit the Progression of Hepatocellular Carcinoma (Review). Oncol. Lett. 2019, 18, 4377–4384.
- Zhang, J.; Liu, Z.; Jia, J. Mechanisms of Smoothened Regulation in Hedgehog Signaling. Cells 2021, 10, 2138.
- Jamieson, C.; Martinelli, G.; Papayannidis, C.; Cortes, J.E. Hedgehog Pathway Inhibitors: A New Therapeutic Class for the Treatment of Acute Myeloid Leukemia. Blood Cancer Discov. 2020, 1, 134–145.
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; Ruiz I Altaba, A. Human Colon Cancer Epithelial Cells Harbour Active HEDGEHOG-GLI Signalling That Is Essential for Tumour Growth, Recurrence, Metastasis and Stem Cell Survival and Expansion. EMBO Mol. Med. 2009, 1, 338–351.
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 Signaling Regulates Human Glioma Growth, Cancer Stem Cell Self-Renewal, and Tumorigenicity. Curr. Biol. 2007, 17, 165–172.
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz Altaba, A. Melanomas Require HEDGEHOG-GLI Signaling Regulated by Interactions between GLI1 and the RAS-MEK/AKT Pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900.
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes de Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread Requirement for Hedgehog Ligand Stimulation in Growth of Digestive Tract Tumours. Nature 2003, 425, 846–851.
- Thayer, S.P.; Pasca di Magliano, M.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernandez-del Castillo, C.; Yajnik, V.; et al. Hedgehog Is an Early and Late of Pancreatic Cancer. Nature 2003, 425, 851–856.
- Feldmann, G.; Dhara, S.; Fendrich, V.; Bedja, D.; Beaty, R.; Mullendore, M.; Karikari, C.; Alvarez, H.; Iacobuzio-Donahue, C.; Jimeno, A.; et al. Blockade of Hedgehog Signaling Inhibits Pancreatic Cancer Invasion and Metastases: A New Paradigm for Combination Therapy in Solid Cancers. Cancer Res. 2007, 67, 2187–2196.
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog Signalling in Prostate, Neoplasia And. Nature 2004, 431, 707–7112.
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog Signalling within Airway Epithelial Progenitors and in Small-Cell Lung Cancer. Nature 2003, 422, 313–317.
- Berman, D.M.; Karhadkar, S.S.; Hallahan, A.R.; Pritchard, J.I.; Eberhart, C.G.; Watkins, D.N.; Chen, J.K.; Cooper, M.K.; Taipale, J.; Olson, J.M.; et al. Medulloblastoma Growth Inhibition by Hedgehog Pathway Blockade. Science (1979) 2002, 297, 1559–1561.
- Sanchez, P.; Ruiz I Altaba, A. In Vivo Inhibition of Endogenous Brain Tumors through Systemic Interference of Hedgehog Signaling in Mice. Mech. Dev. 2005, 122, 223–230.
- Chen, J.K. I Only Have Eye for Ewe: The Discovery of Cyclopamine and Development of Hedgehog Pathway-Targeting Drugs. Nat. Prod. Rep. 2016, 33, 595–601.
- Cho, W.; Kong, H.; Gadeau, A.-P.; Sharma, K.; Sasai, N.; Jp, N.N.; Toriyama, M.; Kondo, T. Hedgehog Signal and Genetic Disorders. Front. Genet. 2019, 10, 1103.
- ClinicalTrials.gov. Study of Patidegib Topical Gel, 2%, for the Reduction of Disease Burden of Persistently Developing Basal Cell Carcinomas (BCCs) in Subjects with Basal Cell Nevus Syndrome (Gorlin Syndrome)—No Study Results Posted. Available online: https://clinicaltrials.gov/ct2/show/results/NCT03703310 (accessed on 20 September 2022).
- FDA. 2012 Notifications. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/2012-notifications (accessed on 24 July 2021).
- FDA. Novel Drug Approvals for 2015. Available online: https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2015 (accessed on 24 July 2021).
- Dummer, R.; Guminski, A.; Gutzmer, R.; Dirix, L.; Lewis, K.D.; Combemale, P.; Herd, R.M.; Kaatz, M.; Loquai, C.; Stratigos, A.J.; et al. The 12-Month Analysis from Basal Cell Carcinoma Outcomes with LDE225 Treatment (BOLT): A Phase II, Randomized, Double-Blind Study of Sonidegib in Patients with Advanced Basal Cell Carcinoma. J. Am. Acad. Dermatol. 2016, 75, 113–125.e5.
- Fania, L.; Didona, D.; Morese, R.; Campana, I.; Coco, V.; di Pietro, F.R.; Ricci, F.; Pallotta, S.; Candi, E.; Abeni, D.; et al. Basal Cell Carcinoma: From Pathophysiology to Novel Therapeutic Approaches. Biomedicines 2020, 8, 449.
- Wolska-Washer, A.; Robak, T. Glasdegib in the Treatment of Acute Myeloid Leukemia. Future Oncol. 2019, 15, 3219–3232.
- Turner, M. Comprehensive Investigation of Bioactive Steroidal Alkaloids in Veratrum Californicum; Boise State University: Boise, ID, USA, 2019.
This entry is offline, you can click here to edit this entry!