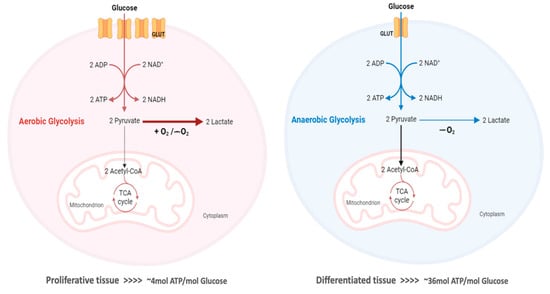
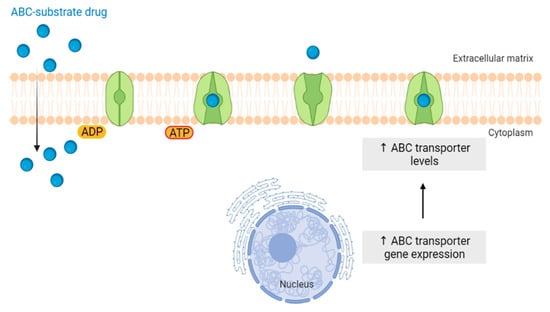
The “Warburg effect” consists of a metabolic shift in energy production from oxidative phosphorylation to glycolysis. The continuous activation of glycolysis in cancer cells causes rapid energy production and an increase in lactate, leading to the acidification of the tumour microenvironment, chemo- and radioresistance, as well as poor patient survival. Nevertheless, the mitochondrial metabolism can be also involved in aggressive cancer characteristics. The metabolic differences between cancer and normal tissues can be considered the Achilles heel of cancer, offering a strategy for new therapies. One of the main causes of treatment resistance consists of the increased expression of efflux pumps, and multidrug resistance (MDR) proteins, which are able to export chemotherapeutics out of the cell. Cells expressing MDR proteins require adenosine triphosphate (ATP) to mediate the efflux of their drug substrates. Thus, inhibition of the main energy-producing pathways in cancer cells, not only induces cancer cell death per se, but also overcomes multidrug resistance.
- tumor microenvironment
- tumor metabolism
- glycolysis
- Warburg effect
- resistance
1. Introduction
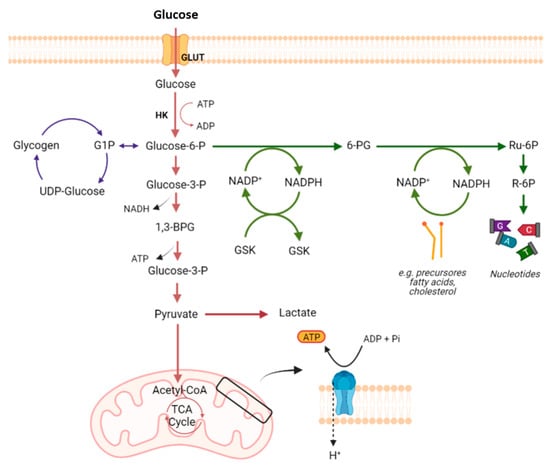
2. Glucose Metabolism
3. The Warburg Effect
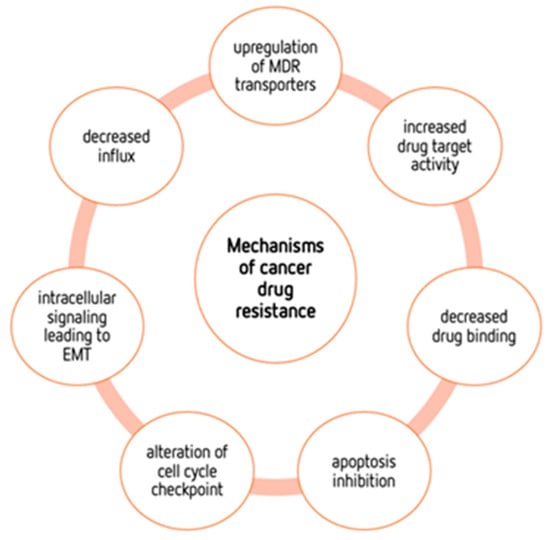
4. Mechanisms of Cancers’ Drug Resistance
4.1. ABC Transporters
4.2. Metabolic Alterations Involved in Drug Resistance in Cancer
4.3. Metabolic Modulation as an Approach to Overcome Drug Resistance
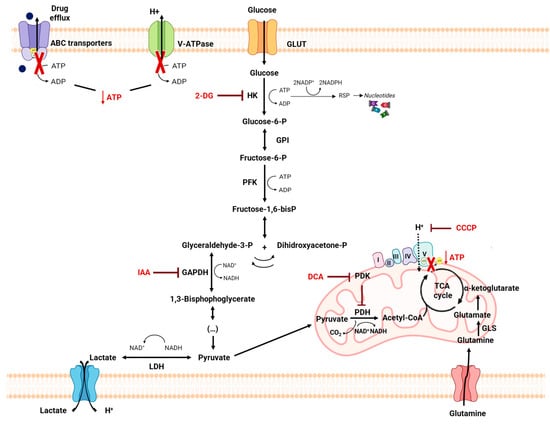
4.4. Self-Delivery of Nanomedicine to Overcome Drug Resistance
5. Conclusions
Although conventional chemotherapy is particularly toxic to tumor cells, it is often non-specific, and is responsible for the significant side effects associated with cancer treatment. However, there are differences between cancer cells and healthy cells that can be explored to increase treatment specificity against cancer. One of these differences consists of the “Warburg effect”, currently considered an emergent cancer hallmark, whereby the upregulation of the glycolytic rate in tumor cells is a key player in acid-resistant phenotypes through their adaptation to hypoxia and acidosis, as well as in tumor aggressiveness [2][9][76]. High glycolytic rates are widely reported to promote the chemoresistance of tumor cells to conventional therapy [2]. In fact, increased acidification of the extracellular space leads to lower drug stability and, consequently, lower drug efficacy. In parallel, the increased production of glycolytic intermediates promotes cell proliferation, since these are biosynthetic precursors, whereas ATP production sustains the activity of proteins involved in both drug efflux and cell division. Together, these effects underly multidrug resistance. Nevertheless, many cancer cells adapt to changes in TME, exhibiting metabolic plasticity and switching their metabolism from glycolysis to OXPHOS, and vice-versa. For example, OXPHOS could be the predominant metabolic pathway used by cancer stem cells, and is often involved in cancer resistance, metastasis, and tumor relapse [77]. Exploring specific characteristics of cancer cells, such as this change in metabolism, could be a promising strategy for the use of more effective and more specific drugs that primarily target cancer cells. In fact, metabolic changes in cancer cells can reveal specific vulnerabilities that could be targeted with precision therapies. However, the metabolic plasticity and interchange of glycolytic and oxidative cells, although occurring many times in the same cancer and being responsible for tumor heterogeneity, is not taken into account in cancer therapies. Thus, more integrated research is needed, investigating the main metabolic pathways used in different conditions and stages of each cancer type, and the influence of the TME characteristics (e.g., oxygen, pH, nutrients availability, immune components) on such metabolic adaptation and heterogeneity. An understanding of these metabolic switches, the identification of metabolic targets, and the use of combined therapies in a more targeted way through the use of nanoparticles could have a huge impact not only on the development of new drugs, but also on the ability to overcome drug resistance, one of the major problems that occurs during cancer treatment.
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics15112610
References
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280.
- Zaal, E.A.; Berkers, C.R. The Influence of Metabolism on Drug Response in Cancer. Front. Oncol. 2018, 8, 500.
- Chen, X.; Chen, S.; Yu, D. Metabolic Reprogramming of Chemoresistant Cancer Cells and the Potential Significance of Metabolic Regulation in the Reversal of Cancer Chemoresistance. Metabolites 2020, 10, 289.
- Ortega, A.D.; Sanchez-Arago, M.; Giner-Sanchez, D.; Sanchez-Cenizo, L.; Willers, I.; Cuezva, J.M. Glucose avidity of carcinomas. Cancer Lett. 2009, 276, 125–135.
- Vanhove, K.; Graulus, G.J.; Mesotten, L.; Thomeer, M.; Derveaux, E.; Noben, J.P.; Guedens, W.; Adriaensens, P. The Metabolic Landscape of Lung Cancer: New Insights in a Disturbed Glucose Metabolism. Front. Oncol. 2019, 9, 1215.
- Cameron, M.E.; Yakovenko, A.; Trevino, J.G. Glucose and Lactate Transport in Pancreatic Cancer: Glycolytic Metabolism Revisited. J. Oncol. 2018, 2018, 6214838.
- Reckzeh, E.S.; Waldmann, H. Small-Molecule Inhibition of Glucose Transporters GLUT-1-4. Chembiochem 2020, 21, 45–52.
- Holman, G.D. Structure, function and regulation of mammalian glucose transporters of the SLC2 family. Pflug. Arch. 2020, 472, 1155–1175.
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314.
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218.
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418.
- Gillies, R.J.; Robey, I.; Gatenby, R.A. Causes and consequences of increased glucose metabolism of cancers. J. Nucl. Med. 2008, 49 (Suppl. S2), 24S–42S.
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516.
- Sun, H.; Zhu, A.; Zhou, X.; Wang, F. Suppression of pyruvate dehydrogenase kinase-2 re-sensitizes paclitaxel-resistant human lung cancer cells to paclitaxel. Oncotarget 2017, 8, 52642–52650.
- Danhier, P.; Banski, P.; Payen, V.L.; Grasso, D.; Ippolito, L.; Sonveaux, P.; Porporato, P.E. Cancer metabolism in space and time: Beyond the Warburg effect. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 556–572.
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757.
- Ghanbari Movahed, Z.; Rastegari-Pouyani, M.; Mohammadi, M.H.; Mansouri, K. Cancer cells change their glucose metabolism to overcome increased ROS: One step from cancer cell to cancer stem cell? Biomed. Pharmacother. 2019, 112, 108690.
- Nadzialek, S.; Vanparys, C.; Van der Heiden, E.; Michaux, C.; Brose, F.; Scippo, M.L.; De Coen, W.; Kestemont, P. Understanding the gap between the estrogenicity of an effluent and its real impact into the wild. Sci. Total Environ. 2010, 408, 812–821.
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482.
- Jesser, E.A.; Brady, N.J.; Huggins, D.N.; Witschen, P.M.; O’Connor, C.H.; Schwertfeger, K.L. STAT5 is activated in macrophages by breast cancer cell-derived factors and regulates macrophage function in the tumor microenvironment. Breast Cancer Res. 2021, 23, 104.
- Putney, L.K.; Barber, D.L. Expression profile of genes regulated by activity of the Na-H exchanger NHE1. BMC Genom. 2004, 5, 46.
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157.
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825.
- Fan, T.W.; Kucia, M.; Jankowski, K.; Higashi, R.M.; Ratajczak, J.; Ratajczak, M.Z.; Lane, A.N. Rhabdomyosarcoma cells show an energy producing anabolic metabolic phenotype compared with primary myocytes. Mol. Cancer 2008, 7, 79.
- Minemura, H.; Takagi, K.; Sato, A.; Yamaguchi, M.; Hayashi, C.; Miki, Y.; Harada-Shoji, N.; Miyashita, M.; Sasano, H.; Suzuki, T. Isoforms of IDH in breast carcinoma: IDH2 as a potent prognostic factor associated with proliferation in estrogen-receptor positive cases. Breast Cancer 2021, 28, 915–926.
- Rezayatmand, H.; Razmkhah, M.; Razeghian-Jahromi, I. Drug resistance in cancer therapy: The Pandora’s Box of cancer stem cells. Stem Cell Res. Ther. 2022, 13, 181.
- De Las Rivas, J.; Brozovic, A.; Izraely, S.; Casas-Pais, A.; Witz, I.P.; Figueroa, A. Cancer drug resistance induced by EMT: Novel therapeutic strategies. Arch. Toxicol. 2021, 95, 2279–2297.
- Fletcher, J.I.; Haber, M.; Henderson, M.J.; Norris, M.D. ABC transporters in cancer: More than just drug efflux pumps. Nat. Rev. Cancer 2010, 10, 147–156.
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309.
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212.
- Jia, D.; Park, J.H.; Kaur, H.; Jung, K.H.; Yang, S.; Tripathi, S.; Galbraith, M.; Deng, Y.; Jolly, M.K.; Kaipparettu, B.A.; et al. Towards decoding the coupled decision-making of metabolism and epithelial-to-mesenchymal transition in cancer. Br. J. Cancer 2021, 124, 1902–1911.
- Inoue, A.; Seidel, M.G.; Wu, W.; Kamizono, S.; Ferrando, A.A.; Bronson, R.T.; Iwasaki, H.; Akashi, K.; Morimoto, A.; Hitzler, J.K.; et al. Slug, a highly conserved zinc finger transcriptional repressor, protects hematopoietic progenitor cells from radiation-induced apoptosis in vivo. Cancer Cell 2002, 2, 279–288.
- Olmeda, D.; Moreno-Bueno, G.; Flores, J.M.; Fabra, A.; Portillo, F.; Cano, A. SNAI1 is required for tumor growth and lymph node metastasis of human breast carcinoma MDA-MB-231 cells. Cancer Res. 2007, 67, 11721–11731.
- Tavares-Valente, D.; Cannone, S.; Greco, M.R.; Carvalho, T.M.A.; Baltazar, F.; Queiros, O.; Agrimi, G.; Reshkin, S.J.; Cardone, R.A. Extracellular Matrix Collagen I Differentially Regulates the Metabolic Plasticity of Pancreatic Ductal Adenocarcinoma Parenchymal Cell and Cancer Stem Cell. Cancers 2023, 15, 3868.
- Yan, L.; Tu, B.; Yao, J.; Gong, J.; Carugo, A.; Bristow, C.A.; Wang, Q.; Zhu, C.; Dai, B.; Kang, Y.; et al. Targeting Glucose Metabolism Sensitizes Pancreatic Cancer to MEK Inhibition. Cancer Res. 2021, 81, 4054–4065.
- Guo, J.; Satoh, K.; Tabata, S.; Mori, M.; Tomita, M.; Soga, T. Reprogramming of glutamine metabolism via glutamine synthetase silencing induces cisplatin resistance in A2780 ovarian cancer cells. BMC Cancer 2021, 21, 174.
- Guo, J.; Yu, J.; Peng, F.; Li, J.; Tan, Z.; Chen, Y.; Rao, T.; Wang, Y.; Peng, J.; Zhou, H. In vitro and in vivo analysis of metabolites involved in the TCA cycle and glutamine metabolism associated with cisplatin resistance in human lung cancer. Expert Rev. Proteom. 2021, 18, 233–240.
- Lotz, C.; Kelleher, D.K.; Gassner, B.; Gekle, M.; Vaupel, P.; Thews, O. Role of the tumor microenvironment in the activity and expression of the p-glycoprotein in human colon carcinoma cells. Oncol. Rep. 2007, 17, 239–244.
- Skeberdyte, A.; Sarapiniene, I.; Aleksander-Krasko, J.; Stankevicius, V.; Suziedelis, K.; Jarmalaite, S. Dichloroacetate and Salinomycin Exert a Synergistic Cytotoxic Effect in Colorectal Cancer Cell Lines. Sci. Rep. 2018, 8, 17744.
- Xiao, H.; Zheng, Y.; Ma, L.; Tian, L.; Sun, Q. Clinically-Relevant ABC Transporter for Anti-Cancer Drug Resistance. Front. Pharmacol. 2021, 12, 648407.
- Robert, J.; Morvan, V.L.; Smith, D.; Pourquier, P.; Bonnet, J. Predicting drug response and toxicity based on gene polymorphisms. Crit. Rev. Oncol. Hematol. 2005, 54, 171–196.
- Aye, I.L.; Singh, A.T.; Keelan, J.A. Transport of lipids by ABC proteins: Interactions and implications for cellular toxicity, viability and function. Chem. Biol. Interact. 2009, 180, 327–339.
- Bugde, P.; Biswas, R.; Merien, F.; Lu, J.; Liu, D.X.; Chen, M.; Zhou, S.; Li, Y. The therapeutic potential of targeting ABC transporters to combat multi-drug resistance. Expert Opin. Ther. Targets 2017, 21, 511–530.
- Wang, S.A.; Young, M.J.; Wang, Y.C.; Chen, S.H.; Liu, C.Y.; Lo, Y.A.; Jen, H.H.; Hsu, K.C.; Hung, J.J. USP24 promotes drug resistance during cancer therapy. Cell Death Differ. 2021, 28, 2690–2707.
- Fletcher, J.I.; Williams, R.T.; Henderson, M.J.; Norris, M.D.; Haber, M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist. Updates 2016, 26, 1–9.
- Welte, Y.; Adjaye, J.; Lehrach, H.R.; Regenbrecht, C.R. Cancer stem cells in solid tumors: Elusive or illusive? Cell Commun. Signal. 2010, 8, 6.
- Scharenberg, C.W.; Harkey, M.A.; Torok-Storb, B. The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood 2002, 99, 507–512.
- Zochbauer-Muller, S.; Filipits, M.; Rudas, M.; Brunner, R.; Krajnik, G.; Suchomel, R.; Schmid, K.; Pirker, R. P-glycoprotein and MRP1 expression in axillary lymph node metastases of breast cancer patients. Anticancer Res. 2001, 21, 119–124.
- Juan-Carlos, P.M.; Perla-Lidia, P.P.; Stephanie-Talia, M.M.; Monica-Griselda, A.M.; Luz-Maria, T.E. ABC transporter superfamily. An updated overview, relevance in cancer multidrug resistance and perspectives with personalized medicine. Mol. Biol. Rep. 2021, 48, 1883–1901.
- Maher, J.C.; Wangpaichitr, M.; Savaraj, N.; Kurtoglu, M.; Lampidis, T.J. Hypoxia-inducible factor-1 confers resistance to the glycolytic inhibitor 2-deoxy-D-glucose. Mol. Cancer Ther. 2007, 6, 732–741.
- Reina-Campos, M.; Moscat, J.; Diaz-Meco, M. Metabolism shapes the tumor microenvironment. Curr. Opin. Cell Biol. 2017, 48, 47–53.
- Liu, C.; Jin, Y.; Fan, Z. The Mechanism of Warburg Effect-Induced Chemoresistance in Cancer. Front. Oncol. 2021, 11, 698023.
- Tavares-Valente, D.; Sousa, B.; Schmitt, F.; Baltazar, F.; Queiros, O. Disruption of pH Dynamics Suppresses Proliferation and Potentiates Doxorubicin Cytotoxicity in Breast Cancer Cells. Pharmaceutics 2021, 13, 242.
- Li, X.; Zhong, Y.; Lu, J.; Axcrona, K.; Eide, L.; Syljuasen, R.G.; Peng, Q.; Wang, J.; Zhang, H.; Goscinski, M.A.; et al. MtDNA depleted PC3 cells exhibit Warburg effect and cancer stem cell features. Oncotarget 2016, 7, 40297–40313.
- Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056.
- Pranzini, E.; Pardella, E.; Paoli, P.; Fendt, S.M.; Taddei, M.L. Metabolic Reprogramming in Anticancer Drug Resistance: A Focus on Amino Acids. Trends Cancer 2021, 7, 682–699.
- Li, T.; Copeland, C.; Le, A. Glutamine Metabolism in Cancer. Adv. Exp. Med. Biol. 2021, 1311, 17–38.
- Ma, W.W.; Jacene, H.; Song, D.; Vilardell, F.; Messersmith, W.A.; Laheru, D.; Wahl, R.; Endres, C.; Jimeno, A.; Pomper, M.G.; et al. fluorodeoxyglucose positron emission tomography correlates with Akt pathway activity but is not predictive of clinical outcome during mTOR inhibitor therapy. J. Clin. Oncol. 2009, 27, 2697–2704.
- Martins, J.P.; das Neves, J.; de la Fuente, M.; Celia, C.; Florindo, H.; Gunday-Tureli, N.; Popat, A.; Santos, J.L.; Sousa, F.; Schmid, R.; et al. The solid progress of nanomedicine. Drug Deliv. Transl. Res. 2020, 10, 726–729.
- Yang, Y.; Zheng, X.; Chen, L.; Gong, X.; Yang, H.; Duan, X.; Zhu, Y. Multifunctional Gold Nanoparticles in Cancer Diagnosis and Treatment. Int. J. Nanomed. 2022, 17, 2041–2067.
- Yao, W.; Yao, J.; Qian, F.; Que, Z.; Yu, P.; Luo, T.; Zheng, D.; Zhang, Z.; Tian, J. Paclitaxel-loaded and folic acid-modified PLGA nanomedicine with glutathione response for the treatment of lung cancer. Acta Biochim. Biophys. Sin. 2021, 53, 1027–1036.
- van den Boogaard, W.M.C.; Komninos, D.S.J.; Vermeij, W.P. Chemotherapy Side-Effects: Not All DNA Damage Is Equal. Cancers 2022, 14, 627.
- Sousa, A.R.; Oliveira, M.J.; Sarmento, B. Impact of CEA-targeting Nanoparticles for Drug Delivery in Colorectal Cancer. J. Pharmacol. Exp. Ther. 2019, 370, 657–670.
- Cunha, A.; Rocha, A.C.; Barbosa, F.; Baiao, A.; Silva, P.; Sarmento, B.; Queiros, O. Glycolytic Inhibitors Potentiated the Activity of Paclitaxel and Their Nanoencapsulation Increased Their Delivery in a Lung Cancer Model. Pharmaceutics 2022, 14, 2021.
- Barenholz, Y. Doxil(R)—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134.
- Rezvantalab, S.; Drude, N.I.; Moraveji, M.K.; Guvener, N.; Koons, E.K.; Shi, Y.; Lammers, T.; Kiessling, F. PLGA-Based Nanoparticles in Cancer Treatment. Front. Pharmacol. 2018, 9, 1260.
- Lu, L.; Peter, S.J.; Lyman, M.D.; Lai, H.L.; Leite, S.M.; Tamada, J.A.; Uyama, S.; Vacanti, J.P.; Langer, R.; Mikos, A.G. In vitro and in vivo degradation of porous poly(DL-lactic-co-glycolic acid) foams. Biomaterials 2000, 21, 1837–1845.
- Luderer, F.; Lobler, M.; Rohm, H.W.; Gocke, C.; Kunna, K.; Kock, K.; Kroemer, H.K.; Weitschies, W.; Schmitz, K.P.; Sternberg, K. Biodegradable sirolimus-loaded poly(lactide) nanoparticles as drug delivery system for the prevention of in-stent restenosis in coronary stent application. J. Biomater. Appl. 2011, 25, 851–875.
- Cai, J.; Qian, K.; Zuo, X.; Yue, W.; Bian, Y.; Yang, J.; Wei, J.; Zhao, W.; Qian, H.; Liu, B. PLGA nanoparticle-based docetaxel/LY294002 drug delivery system enhances antitumor activities against gastric cancer. J. Biomater. Appl. 2019, 33, 1394–1406.
- Zhang, L.; Zhai, B.Z.; Wu, Y.J.; Wang, Y. Recent progress in the development of nanomaterials targeting multiple cancer metabolic pathways: A review of mechanistic approaches for cancer treatment. Drug Deliv. 2023, 30, 1–18.
- Ren, M.; Zheng, X.; Gao, H.; Jiang, A.; Yao, Y.; He, W. Nanomedicines Targeting Metabolism in the Tumor Microenvironment. Front. Bioeng. Biotechnol. 2022, 10, 943906.
- Sa, P.; Sahoo, S.K.; Dilnawaz, F. Responsive Role of Nanomedicine in the Tumor Microenvironment and Cancer Drug Resistance. Curr. Med. Chem. 2023, 30, 3335–3355.
- Sasaki, K.; Nishina, S.; Yamauchi, A.; Fukuda, K.; Hara, Y.; Yamamura, M.; Egashira, K.; Hino, K. Nanoparticle-Mediated Delivery of 2-Deoxy-D-Glucose Induces Antitumor Immunity and Cytotoxicity in Liver Tumors in Mice. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 739–762.
- Yang, B.; Chen, Y.; Shi, J. Tumor-Specific Chemotherapy by Nanomedicine-Enabled Differential Stress Sensitization. Angew. Chem. Int. Ed. Engl. 2020, 59, 9693–9701.
- Guimaraes, P.P.G.; Gaglione, S.; Sewastianik, T.; Carrasco, R.D.; Langer, R.; Mitchell, M.J. Nanoparticles for Immune Cytokine TRAIL-Based Cancer Therapy. ACS Nano 2018, 12, 912–931.
- Akins, N.S.; Nielson, T.C.; Le, H.V. Inhibition of Glycolysis and Glutaminolysis: An Emerging Drug Discovery Approach to Combat Cancer. Curr. Top. Med. Chem. 2018, 18, 494–504.
- Liu, G.; Luo, Q.; Li, H.; Liu, Q.; Ju, Y.; Song, G. Increased Oxidative Phosphorylation Is Required for Stemness Maintenance in Liver Cancer Stem Cells from Hepatocellular Carcinoma Cell Line HCCLM3 Cells. Int. J. Mol. Sci. 2020, 21, 5276.