Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Mitochondria are essential organelles for energy production, calcium homeostasis, redox signaling, and other cellular responses involved in pulmonary vascular biology and disease processes. Mitochondrial homeostasis depends on a balance in mitochondrial fusion and fission (dynamics). Mitochondrial dynamics are regulated by a viable circadian clock.
- mitochondria
- circadian molecules
- nicotine
- pulmonary vascular dysfunction
1. Introduction
Mitochondria have for a long time been identified as being the “powerhouse” of the cell, where the production of energy provides the entire functioning of cellular metabolism. However, mitochondria are much more than that, they can act in several other biological processes. In the last few decades, research advancements have revolutionized our comprehension of multifaceted and intricate mitochondrial functions. These complex organelles have emerged as central players in diverse biological processes, including redox signaling, calcium homeostasis, apoptosis, autophagy, and other cellular responses. Moreover, cutting-edge imaging techniques have facilitated a thorough investigation of mitochondrial dynamics, which control the regulation of mitochondrial number and size via fusion and fission processes, ultimately offering a fascinating and perplexing glimpse into the inner workings of these organelles. The balance between these two processes is particularly important, as they control and regulate their formation and distribution, thereby mediating normal cellular functions and disease processes [1][2].
Mitochondrial fusion is the union of two mitochondria in one larger mitochondrion (Figure 1). They unite their outer membranes and then fuse their inner membranes. Each sub-compartment (inner, outer membrane, and matrix) blends with its respective sub-compartment from the other mitochondrion. The main components involved in mitochondrial fusion are also members of the dynamin family of GTPases (mitofusin 1 and 2, MFN1/2) in the outer membrane, and optic atrophy 1 (OPA1) in the inner membrane. MFN1/2 facilitate external membrane fixation and subsequent fusion [3][4]. Thus, mitochondrial fusion is considered beneficial as it is associated with an increase in mitochondrial function and ATP production.
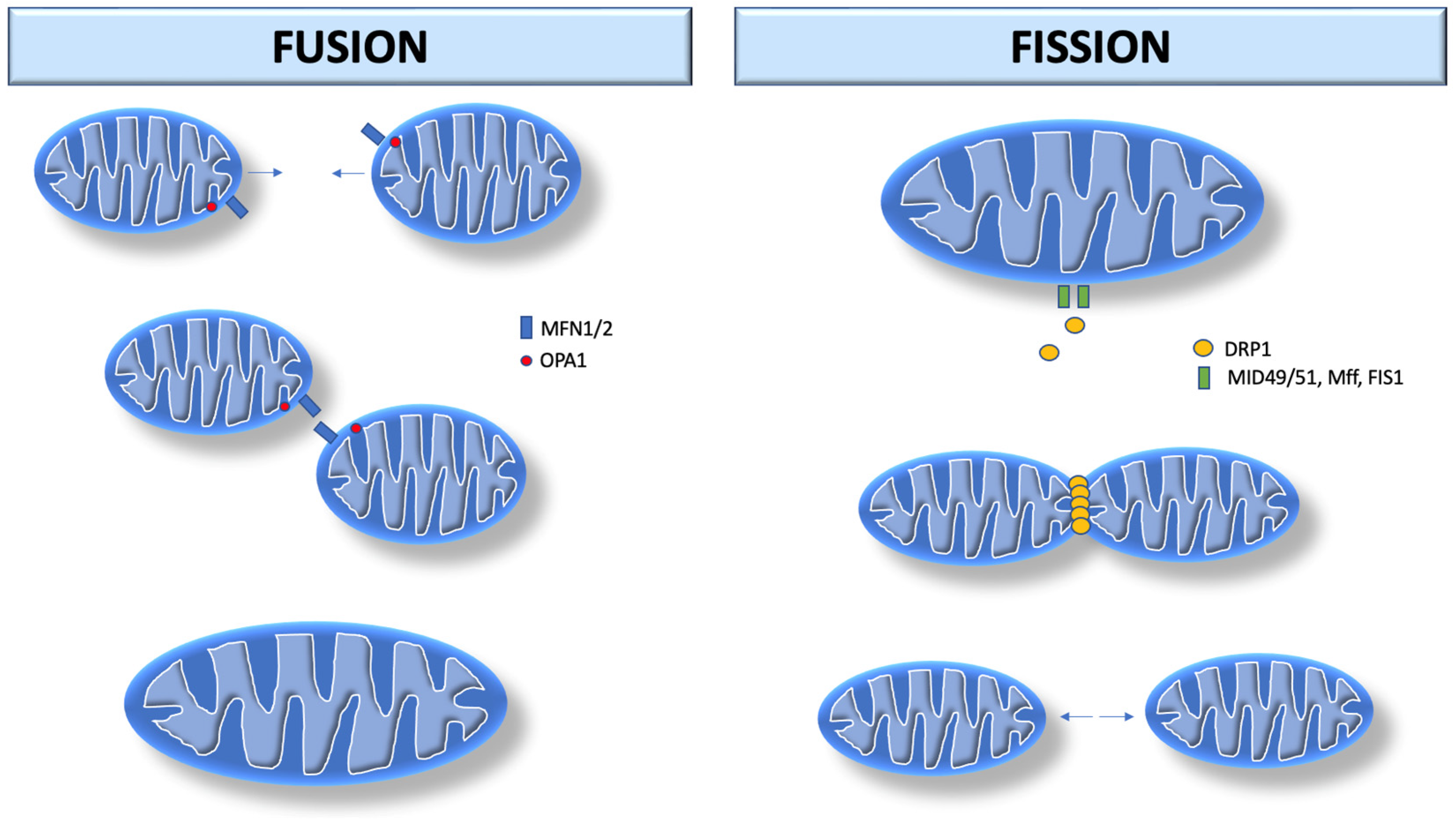
Figure 1. Mitochondrial dynamics. Mitochondrial fusion begins with MFN1/2 in the outer membrane, and OPA1 in the inner membrane. MFN1/2 facilitates external membrane fixation and subsequent fusion. Mitochondrial fission begins after recruitment of DRP1 by MiD49/MiD51. DRP1 performs fission by self-polymerization around the mitochondrial outer membrane, where it contracts the organelle in a process that uses GTP hydrolysis. MFN1/2: Mitofusin 1 and 2; OPA-1: optic atrophy 1. DRP1: dynamin-related protein 1; MiD49: mitochondrial dynamics protein 49 kDA; MiD51: mitochondrial dynamics protein 51 kDA; Mff: mitochondrial fission factor; FIS1: fission protein 1.
Mitochondrial fission consists of the division of one mitochondrion into two separate organelles. The primary mediator of mitochondrial fission is dynamin-related protein 1 (DRP1, encoded by DNM1L in humans), a GTPase that is recruited to the mitochondrial outer membrane from the cytosol by binding to mitochondrial fission factor adapter proteins as well as 49 and 51 kDa mitochondrial dynamics proteins (MiD49/MiD51). After recruitment into the mitochondria, DRP1 performs fission by self-polymerization around the mitochondrial outer membrane, where it contracts the organelle in a process via GTP hydrolysis. Once assembled, DRP1 is sufficient to perform membrane constriction and cutting [3][5]. In contrast to mitochondrial fusion, mitochondrial fission appears to be harmful as it is associated with decreased mitochondrial functions and increased reactive oxygen species (ROS) production [6][7]. Damaged mitochondria produce more superoxide anion [8] and hydrogen peroxide [9], which potentiate mitochondrial ROS propagation [10].
Mitochondrial morphology can vary dramatically between cells and tissues. Deregulation of mitochondrial fission and fusion results in a fragmented network characterized by many small round-shaped mitochondria or hyperfused mitochondria with elongated and highly connected shapes. These balanced dynamic transitions are necessary to ensure normal mitochondrial functions and respond to cellular needs, adapting the network to nutrient availability and cell metabolic state [11].
2. Role of Mitochondrial Fission in Pulmonary Hypertension
Mitochondrial fusion allows an exchange of material between the mitochondria, while fission allows for intact mitochondria to separate. Nutrient deprivation modulates mitochondrial morphology, thus stimulating mitochondrial fusion [12], while nutrient overload is often associated with mitochondrial fission [13]. Mitochondrial fusion contributes to the formation of an interconnected mitochondrial matrix, whereas mitochondrial fission leads to the formation of smaller mitochondria that are connected to each other or the endoplasmic reticulum. Several factors contribute to the regulation of mitochondrial fission and fusion, including cell-cycle kinases [14][15], phosphatases [16], cellular redox enzymes, intracellular calcium levels, calcium-dependent kinases [17], and metabolism. Mitochondrial dynamics are strongly linked to cell proliferation, apoptosis, and mitochondrial quality [18]. Both fission and fusion are also associated with various diseases including PH [19].
PH is defined as a mean pulmonary artery pressure (mPAP) ≥ 25 mmHg at rest [20][21]. The World Health Organization (WHO) has classified PH into the following five groups based on their origin, prognosis, and management: Group 1 mainly includes idiopathic or family pulmonary arterial hypertension; Group 2 merely occurs due to left heart disease; Group 3 is due to chronic lung disease (e.g., chronic obstructive pulmonary disease, interstitial lung disease, or overlap syndromes) or conditions that cause hypoxia (e.g., obstructive sleep apnea, alveolar hypoventilation disorders); Group 4 is called chronic thromboembolic pulmonary hypertension (CTEPH). CTEPH can occur when the body is not able to dissolve a blood clot in the lungs. This can lead to scar tissue in the blood vessels of the lungs, which blocks normal blood flow and makes the right side of the heart work harder; and Group 5 is a heterogeneous group of diseases that encompass PH secondary to multifactorial mechanisms, including sickle cell disease and sarcoidosis. Among these, PH attributable to left heart and lung diseases are the second most prevalent subtype with an estimated prevalence of up to 50% [22].
The enigmatic pathogenesis linking the mitochondrial dynamics to PH remains a challenge worldwide. Recent investigations have suggested the putative involvement of high-mobility group box-1 (HMGB1) and aberrant mitochondrial fission, resulting from the unwarranted activation of the DRP1, in patients with PH. Nonetheless, it remains a mystery whether DRP1-driven mitochondrial fission or its downstream targets provokes the HMGB1-induced migration and proliferation of PASMC in PH [23]. Further research to address this question is needed to better elucidate the mechanistic underpinnings of PH.
Changes in mitochondrial morphology have also been implicated in mitochondrial Ca2+ regulation and ROS production. ROS generation leads to inflammation and autoimmunity, which are important factors in the etiology of PH [24]. Mitochondrial morphology acts by regulating mitochondrial Ca2+ uptake to impact cellular Ca2+ homeostasis. Increasing fusion due to DRP1 knockdown increases the mitochondrial Ca2+ retention capacity. On the other hand, increasing fission because of MFN2 knockdown strongly reduces mitochondrial Ca2+ [25]. Altered mitochondrial dynamics play a direct role in pulmonary vascular dysfunction in PH through various switches such as increased oxidative stress and inflammation [19][26][27].
In PH, acquired mitochondrial abnormalities, including epigenetic silencing of superoxide dismutase (SOD2), disrupt oxygen sensing creating a pseudo-hypoxic environment characterized by normoxic activation of hypoxia-inducible factor-1α (HIF-1α). In addition, altered mitochondrial dynamics result in mitochondrial fragmentation. The molecular basis of this structural change includes the upregulation and activation of fission mediators, notably DRP-1, and the downregulation of MFN2. These pathogenic mitochondrial abnormalities offer new therapeutic targets. Inhibition of mitotic fission or enhancement of fusion in PH PASMC slows cell proliferation, causes cell cycle arrest, and induces apoptosis. DRP-1 inhibition or MFN2 gene therapy can regress PH [28].
3. Mitochondrial Dynamic Dysfunction in Hypoxia Contribute to Group 3 PH
Changes in mitochondrial dynamics are correlated with decreased mitochondrial respiratory enzyme activity and ATP abundance in hypoxic PH. Moreover, PASMCs from PA present dysmorphic mitochondria with reduced respiratory chain coupling, inefficient use of oxygen, and increased glycolysis [29]. These pathological changes were significantly attenuated in knockdown HIF-1α cells. The HIF-1α knockdown also significantly increases the mitochondrial length/width ratio, reducing the number of mitochondria, and the mitochondrial area [29].
After hypoxia stimulation, lung tissues and PASMCs present reduced mitochondrial respiratory complex IV and ATP, increased ROS production, and down-regulated mtDNA content, indicating hypoxia-induced deficiencies in energy metabolism and enzymatic activity of the respiratory complex [29]. Hypoxia also promotes mitochondrial retention in the perinuclear area, partial microtubule dissociation, anomalous fusion activity, and suppression of general fusion activity, leading to mitochondrial shortening. A key trigger for this response is the suppression of mitochondrial ATP production and the depletion of cellular ATP [30].
The expression of the fusion proteins MFN1/2 is significantly reduced in pulmonary artery tissues from rats with PH. In addition, MFN1/2 mRNA levels are significantly decreased in PASMCs after exposure to hypoxia compared to normoxia. These results suggest that mitochondrial fusion proteins MFN1/2 are involved in pulmonary vascular remodeling and PH [29]. The expression levels of the DRP1 fission protein also show a significant increase in PASMCs following hypoxia, both in vivo and in vitro. This suggests that the mitochondrial fission protein DRP1 is involved in pulmonary vascular remodeling. HIF-1α mediates the hypoxic activation of DRP1 to result in the induction of the mitochondrial pathway of proliferation and apoptosis in PASMCs via modulation of protein cell nuclear antigen (PCNA) and caspase-3 expression [29].
Nox4 is a major subunit of NADPH oxidase. Its expression is increased in murine models of hypoxia-induced PH in the pulmonary vasculature of patients with PH [31]. Hypoxia increases Nox4, which promotes the increase of mitochondrial hydrogen peroxide, promoting PH [32]. Nox4 silencing by siRNA causes reduction of ROS levels under normoxic and hypoxic conditions and suppresses the hypoxia-induced significant ROS increase in pulmonary adventitial artery fibroblasts [33].
Studies have also shown that Nox4 is found within mitochondria [34][35]. Nox4 modulates the activity of enzyme complexes within the electron transport chain (ETC) [36], in addition to its interaction with complex I, inhibiting its activity [9]. As hydrogen peroxide (H2O2) is the main product of Nox4 activity, the presence of active Nox4 in mitochondria can be expected to increase mitochondrial H2O2 levels. Koziel et al., demonstrated a significant decrease in the concentration of H2O2 in the mitochondria of Nox4-knockout cells [37]. As another subunit of NADPH oxidase, an important finding regarding Nox4 is its role in mitochondrial morphology, as in Nox4-knockout cells mitochondria revealed a characteristic of highly separated, non-interconnected networks [37]. Furthermore, the increase in mitochondrial Nox4 expression induces vascular SMC-mediated structural remodeling of the vascular wall due to the increase in ROS [38].
PKCε is a cytosolic protein that can be translocated to mitochondria in certain situations, even though little is known about this mechanism [39]. PKC-ε also appears to play an important role in mitochondrial morphology. An interesting study established that PKC-ε activation in renal proximal tubular cells induces mitochondrial dysfunction and fragmentation, energy deficit, ROS generation and cell death [40]. Rieske iron-sulfur protein (RISP) is a primary key factor in the generation of ROS originated from mitochondria. The studies and others reveal that RISP is an important primary molecule for initiating hypoxia-induced [ROS]mito generation in PASMCs, cardiac myocytes, and neuronal cells [41]. Researchers further demonstrate that [ROS]mito can subsequently activate cytosolic PKCε and then cell membrane NOX to induce further ROS generation; this ROS-induced ROS production ultimately cause massive increases in [ROS]i in PASMCs [42][43].
RISP-mediated increase of intracellular ROS may subsequently inhibit voltage-gated potassium (Kv) channels, and activates transient receptor potential (TRP) channels, ryanodine receptors (RyRs) (especially RyR2) as well as inosol 1,4,5-trisphosphate receptors (IP3Rs) to evoke a large increase in [Ca2+]i, leading to numerous cellular responses. It is worth pointing out that RyRs may mediate the hypoxic inhibition of KV channels, activation of TRP channels, and amplify IP3R-dependent Ca2+ release [44][45]. Researchers' very recent investigations discover that RyR2-mediated Ca2+ release from the sarcoplasmic reticulum can further promote mitochondrial ROS generation [42].
The pathophysiologic relevance and therapeutic implications of defective mitochondrial fusion and excessive fission in PH have been widely studied. PASMCs play a critical and central role in the development, progression, and advancement of PH. The mitochondria in PASMCs play a critical role as vascular sensors of oxygen and thus may respond to a slight increment or decrement of oxygen tension in the pulmonary artery by activating hypoxic pulmonary vasoconstriction (HPV) [46][47]. HPV is the mechanism employed by the lungs to rectify any ventilation-perfusion mismatch [48]. A decline in oxygen levels in the pulmonary vasculature gives rise to vasoconstriction and shunts the blood-flow towards promoting better perfusion in well-ventilated lung areas. HPV is also important in optimizing partial pressure of CO2 in certain lung conditions including atelectasis and pneumonia [49]. The PASMCs inside these arterial segments have mitochondria that behave as though they have been exposed to continuous hypoxia, and the pathophysiology of PH is centered in these same resistant arteries. In particular, the mitochondria in PH PASMCs exhibit poor metabolism because of transcriptionally mediated inhibition of mitochondrial pyruvate dehydrogenase and are fragmented as a result of an imbalance between mitochondrial fission and fusion.
The hypothesis known as the “redox hypothesis” proposes that the ROS production from the electron transport chain (ETC) complexes I and III in the mitochondria is altered in direct correlation to the level of alveolar PO2, thereby initiating HPV in the human body [50]. The disruption of electron flow and reduction in levels of the diffusible second messenger H2O2 serve as markers of acute hypoxia [51]. The consensus among experts is that the mitochondria act as a primary oxygen sensor in the body, monitoring the levels of ROS in the system as an indicator of hypoxia. However, some dissenting opinions suggest that there may be a paradoxical increase in ROS levels during hypoxia [52].
A set of scientific data reveal that the synthesis of diffusible redox mediators, including radicals and peroxides, is diminished specifically in the resistance PASMC (as opposed to the conduit artery PASMC) during physiologic hypoxia (as opposed to anoxia). As such, hypoxia may lead to a decrease in the generation of ROS, which, in turn, causes inhibition of Kv channels, membrane depolarization, activation of voltage-gated L-type calcium (Cav1) channels, depolarizing the PASMCs. Vasoconstriction begins because of the calcium entry [48][50][53].
The occurrence of HPV in resistance PAs can be attributed to the distinctive capacity of PASMC mitochondria to regulate their ROS production [54]. This phenomenon is not observed in other arteries such as the renal arteries, where the production of ROS in response to changes in PO2 is not significantly altered, and hypoxia results in arterial dilation instead [54]. The precise role of mitochondrial dynamics in HPV is yet to be fully comprehended. However, mitochondrial fission is an obligatory preliminary step in the mechanisms that precede changes in mitochondrial ETC function and ROS signaling in the ductus arteriosus [55]. Although HPV can cause acute PH and contribute to diseases like high altitude pulmonary edema in genetically predisposed people, it is imperative to understand that the underlying mechanism that leads to PH is ROS generation [56]. Hypoxia suppresses the mitochondrial pathways of ROS generation that underlie the oxygen-sensing function of mitochondria. More specifically, chronic hypoxia activates HIF-1α which in turn decreases mitochondrial H2O2 production and minimizes PO2-sensitive ROS generation. This in turn decreases HPV [57].
PH related to chronic hypoxia may also primarily result from the remodeling of pulmonary vessels (ex. medial hypertrophy of small pulmonary arteries (<200 μm) in addition to HPV [48]. This vasculopathy in PH is caused by abnormalities in redox signaling (activated HIF-1 and decreased SOD2), oxidative metabolism (increased PDK and inhibited PDH), mitochondrial dynamics (increased dynamin related protein 1, DRP1, and reduced MFN2), and effector targets (altered expression of O2-sensing Kv channels). Warburg hypothesis proposes that this impaired O2-sensing impairment contributes to the underlying pathology in PH in a similar pattern like cancer relies on glycolysis despite availability of oxygen availability for oxidative metabolism. This theory suggests that both PH and cancer are reliant on the failure of oxygen-sensing due to the alteration of mitochondrial redox functions. This, in turn, manifests as further impairment of oxygen-sensing which in the longer term gives rise to the Warburg phenomenon [58].
The role of the Cyclin B and CDK1 complex has been investigated to play a role in PH. Cyclin B-dependent CDK1 initiates a cycle of mitosis by phosphorylating DRP1 at serine 616, therefore activating mitochondrial fission [59]. On the other hand, inhibition of mitotic fission arrests the cell-cycle at G2/M transition, promoting cell death [60]. However, whether hypoxic fragmentation of mitochondria leads to vasoconstriction in PH is still largely unknown. In ductus arteriosus, a change in PO2 results in rapid (<60 s) mitochondrial fission, ultimately resulting in ROS production, inflammation, and vasoconstriction [55].
Von-Hippel Lindau disease (VHL), a genetic disease discovered in the Chuvash region of Russia, is a striking example of the impairment of oxygen-sensing in mitochondria as the underlying pathophysiology of pulmonary hypertension. A loss-of-function mutation in the VHL factor gives rise to pulmonary hypertension due to normoxic activation of HIF-1α [61][62]. The spectrum of symptoms in VHL comprises polycythemia and PH despite normal oxygen levels. This phenomenon demonstrates that impaired oxygen sensing (and the resulting normoxic activation of HIF-1α and HIF-2α) is sufficient to cause PH [61].
Mitochondrial redox signaling mechanisms in PH create a transcriptional and proteomic fingerprint like that observed in sustained hypoxia. These abnormalities are seen redundantly despite high-oxygen conditions, such as in cell-culture, which provide a state of pseudohypoxia.
PH-associated abnormalities in the pulmonary vascular oxygen-sensing pathway include persistent activation of HIF-1α during normoxic conditions [63] and a transcriptional activation of enzyme PDK in pulmonary arteries [54][63] and RV [64][65]. This causes a rapid shift from oxidative metabolism to aerobic glycolysis, and in turn, impaired mitochondrial fusion, and impaired fission, which results in the fragmentation of the PASMC’s mitochondrial network [60][66].
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines12010053
References
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459.
- Liesa, M.; Palacín, M.; Zorzano, A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009, 89, 799–845.
- Yapa, N.M.B.; Lisnyak, V.; Reljic, B.; Ryan, M.T. Mitochondrial dynamics in health and disease. FEBS Lett. 2021, 595, 1184–1204.
- Chandhok, G.; Lazarou, M.; Neumann, B. Structure, function, and regulation of mitofusin-2 in health and disease. Biol. Rev. Camb. Philos. Soc. 2018, 93, 933–949.
- Kamerkar, S.C.; Kraus, F.; Sharpe, A.J.; Pucadyil, T.J.; Ryan, M.T. Dynamin-related protein 1 has membrane constricting and severing abilities sufficient for mitochondrial and peroxisomal fission. Nat. Commun. 2018, 9, 5239.
- Meng, T.T.; Wang, W.; Meng, F.L.; Wang, S.Y.; Wu, H.H.; Chen, J.M.; Zheng, Y.; Wang, G.X.; Zhang, M.X.; Li, Y.; et al. Nicotine Causes Mitochondrial Dynamics Imbalance and Apoptosis Through ROS Mediated Mitophagy Impairment in Cardiomyocytes. Front. Physiol. 2021, 12, 650055.
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531.
- Sengupta, S.; Yang, G.; O’Donnell, J.C.; Hinson, M.D.; McCormack, S.E.; Falk, M.J.; La, P.; Robinson, M.B.; Williams, M.L.; Yohannes, M.T.; et al. The circadian gene Rev-erbα improves cellular bioenergetics and provides preconditioning for protection against oxidative stress. Free Radic. Biol. Med. 2016, 93, 177–189.
- Hirschhäuser, C.; Bornbaum, J.; Reis, A.; Böhme, S.; Kaludercic, N.; Menabò, R.; Di Lisa, F.; Boengler, K.; Shah, A.M.; Schulz, R.; et al. NOX4 in Mitochondria: Yeast Two-Hybrid-Based Interaction with Complex I Without Relevance for Basal Reactive Oxygen Species? Antioxid. Redox Signal. 2015, 23, 1106–1112.
- Park, J.; Lee, J.; Choi, C. Mitochondrial network determines intracellular ROS dynamics and sensitivity to oxidative stress through switching inter-mitochondrial messengers. PLoS ONE 2011, 6, e23211.
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117.
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: Regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 2015, 32, 678–692.
- Molina, A.J.; Wikstrom, J.D.; Stiles, L.; Las, G.; Mohamed, H.; Elorza, A.; Walzer, G.; Twig, G.; Katz, S.; Corkey, B.E.; et al. Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes 2009, 58, 2303–2315.
- Mitra, K.; Wunder, C.; Roysam, B.; Lin, G.; Lippincott-Schwartz, J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc. Natl. Acad. Sci. USA 2009, 106, 11960–11965.
- Rehman, J.; Zhang, H.J.; Toth, P.T.; Zhang, Y.; Marsboom, G.; Hong, Z.; Salgia, R.; Husain, A.N.; Wietholt, C.; Archer, S.L. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012, 26, 2175–2186.
- Cribbs, J.T.; Strack, S. Functional characterization of phosphorylation sites in dynamin-related protein 1. Methods Enzymol. 2009, 457, 231–253.
- Han, X.J.; Lu, Y.F.; Li, S.A.; Kaitsuka, T.; Sato, Y.; Tomizawa, K.; Nairn, A.C.; Takei, K.; Matsui, H.; Matsushita, M. CaM kinase I α-induced phosphorylation of Drp1 regulates mitochondrial morphology. J. Cell Biol. 2008, 182, 573–585.
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065.
- Yu, Q.; Chan, S.Y. Mitochondrial and Metabolic Drivers of Pulmonary Vascular Endothelial Dysfunction in Pulmonary Hypertension. Adv. Exp. Med. Biol. 2017, 967, 373–383.
- McLaughlin, V.V.; Archer, S.L.; Badesch, D.B.; Barst, R.J.; Farber, H.W.; Lindner, J.R.; Mathier, M.A.; McGoon, M.D.; Park, M.H.; Rosenson, R.S.; et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: A report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: Developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation 2009, 119, 2250–2294.
- Hoeper, M.M.; Bogaard, H.J.; Condliffe, R.; Frantz, R.; Khanna, D.; Kurzyna, M.; Langleben, D.; Manes, A.; Satoh, T.; Torres, F.; et al. Definitions and diagnosis of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D42–D50.
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 2022, 43, 3618–3731.
- Feng, W.; Wang, J.; Yan, X.; Zhang, Q.; Chai, L.; Wang, Q.; Shi, W.; Chen, Y.; Liu, J.; Qu, Z.; et al. ERK/Drp1-dependent mitochondrial fission contributes to HMGB1-induced autophagy in pulmonary arterial hypertension. Cell Prolif. 2021, 54, e13048.
- Huertas, A.; Perros, F.; Tu, L.; Cohen-Kaminsky, S.; Montani, D.; Dorfmüller, P.; Guignabert, C.; Humbert, M. Immune dysregulation and endothelial dysfunction in pulmonary arterial hypertension: A complex interplay. Circulation 2014, 129, 1332–1340.
- Kowaltowski, A.J.; Menezes-Filho, S.L.; Assali, E.A.; Gonçalves, I.G.; Cabral-Costa, J.V.; Abreu, P.; Miller, N.; Nolasco, P.; Laurindo, F.R.M.; Bruni-Cardoso, A.; et al. Mitochondrial morphology regulates organellar Ca. FASEB J. 2019, 33, 13176–13188.
- Richardson, R.B.; Mailloux, R.J. Mitochondria Need Their Sleep: Redox, Bioenergetics, and Temperature Regulation of Circadian Rhythms and the Role of Cysteine-Mediated Redox Signaling, Uncoupling Proteins, and Substrate Cycles. Antioxidants 2023, 12, 674.
- Wang, Q.; Sundar, I.K.; Lucas, J.H.; Muthumalage, T.; Rahman, I. Molecular clock REV-ERBα regulates cigarette smoke-induced pulmonary inflammation and epithelial-mesenchymal transition. JCI Insight 2021, 6, e145200.
- Ryan, J.; Dasgupta, A.; Huston, J.; Chen, K.H.; Archer, S.L. Mitochondrial dynamics in pulmonary arterial hypertension. J. Mol. Med. 2015, 93, 229–242.
- Chen, X.; Yao, J.M.; Fang, X.; Zhang, C.; Yang, Y.S.; Hu, C.P.; Chen, Q.; Zhong, G.W. Hypoxia promotes pulmonary vascular remodeling via HIF-1α to regulate mitochondrial dynamics. J. Geriatr. Cardiol. 2019, 16, 855–871.
- Liu, X.; Hajnóczky, G. Altered fusion dynamics underlie unique morphological changes in mitochondria during hypoxia–reoxygenation stress. Cell Death Differ. 2011, 18, 1561–1572.
- Mittal, M.; Roth, M.; König, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 2007, 101, 258–267.
- Adesina, S.E.; Kang, B.Y.; Bijli, K.M.; Ma, J.; Cheng, J.; Murphy, T.C.; Michael Hart, C.; Sutliff, R.L. Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension. Free Radic. Biol. Med. 2015, 87, 36–47.
- Li, S.; Tabar, S.S.; Malec, V.; Eul, B.G.; Klepetko, W.; Weissmann, N.; Grimminger, F.; Seeger, W.; Rose, F.; Hänze, J. NOX4 regulates ROS levels under normoxic and hypoxic conditions, triggers proliferation, and inhibits apoptosis in pulmonary artery adventitial fibroblasts. Antioxid. Redox Signal. 2008, 10, 1687–1698.
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390.
- Graham, K.A.; Kulawiec, M.; Owens, K.M.; Li, X.; Desouki, M.M.; Chandra, D.; Singh, K.K. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol. Ther. 2010, 10, 223–231.
- Bernard, K.; Logsdon, N.J.; Miguel, V.; Benavides, G.A.; Zhang, J.; Carter, A.B.; Darley-Usmar, V.M.; Thannickal, V.J. NADPH Oxidase 4 (Nox4) Suppresses Mitochondrial Biogenesis and Bioenergetics in Lung Fibroblasts via a Nuclear Factor Erythroid-derived 2-like 2 (Nrf2)-dependent Pathway. J. Biol. Chem. 2017, 292, 3029–3038.
- Kozieł, R.; Pircher, H.; Kratochwil, M.; Lener, B.; Hermann, M.; Dencher, N.A.; Jansen-Dürr, P. Mitochondrial respiratory chain complex I is inactivated by NADPH oxidase Nox4. Biochem. J. 2013, 452, 231–239.
- Canugovi, C.; Stevenson, M.D.; Vendrov, A.E.; Hayami, T.; Robidoux, J.; Xiao, H.; Zhang, Y.Y.; Eitzman, D.T.; Runge, M.S.; Madamanchi, N.R. Increased mitochondrial NADPH oxidase 4 (NOX4) expression in aging is a causative factor in aortic stiffening. Redox Biol. 2019, 26, 101288.
- Budas, G.R.; Churchill, E.N.; Disatnik, M.H.; Sun, L.; Mochly-Rosen, D. Mitochondrial import of PKCepsilon is mediated by HSP90: A role in cardioprotection from ischaemia and reperfusion injury. Cardiovasc. Res. 2010, 88, 83–92.
- Nowak, G.; Bakajsova, D.; Samarel, A.M. Protein kinase C-epsilon activation induces mitochondrial dysfunction and fragmentation in renal proximal tubules. Am. J. Physiol. Renal Physiol. 2011, 301, F197–F208.
- Korde, A.S.; Yadav, V.R.; Zheng, Y.M.; Wang, Y.X. Primary role of mitochondrial Rieske iron–sulfur protein in hypoxic ROS production in pulmonary artery myocytes. Free Radic. Biol. Med. 2011, 50, 945–952.
- Truong, L.; Zheng, Y.M.; Wang, Y.X. Mitochondrial Rieske iron–sulfur protein in pulmonary artery smooth muscle: A key primary signaling molecule in pulmonary hypertension. Arch. Biochem. Biophys. 2019, 664, 68–75.
- Truong, L.N.; Santos, E.W.; Zheng, Y.M.; Wang, Y.X. Rieske Iron-Sulfur Protein Mediates Pulmonary Hypertension Following Nicotine/Hypoxia Co-Exposure. Am. J. Respir. Cell Mol. Biol. 2023. ahead-of-print.
- Tykocki, N.R.; Boerman, E.M.; Jackson, W.F. Smooth Muscle Ion Channels and Regulation of Vascular Tone in Resistance Arteries and Arterioles. Compr. Physiol. 2017, 7, 485–581.
- Wang, Y.X.; Reyes-García, J.; Di Mise, A.; Zheng, Y.M. Role of ryanodine receptor 2 and FK506-binding protein 12.6 dissociation in pulmonary hypertension. J. Gen. Physiol. 2023, 155, e202213100.
- Weir, E.K.; López-Barneo, J.; Buckler, K.J.; Archer, S.L. Acute oxygen-sensing mechanisms. N. Engl. J. Med. 2005, 353, 2042–2055.
- Archer, S.L.; Huang, J.; Henry, T.; Peterson, D.; Weir, E.K. A redox-based O2 sensor in rat pulmonary vasculature. Circ. Res. 1993, 73, 1100–1112.
- Weir, E.K.; Archer, S.L. The mechanism of acute hypoxic pulmonary vasoconstriction: The tale of two channels. FASEB J. 1995, 9, 183–189.
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P. Hypoxic pulmonary vasoconstriction. Physiol. Rev. 2012, 92, 367–520.
- Archer, S.L.; Will, J.A.; Weir, E.K. Redox status in the control of pulmonary vascular tone. Herz 1986, 11, 127–141.
- Archer, S.L.; Nelson, D.P.; Weir, E.K. Simultaneous measurement of O2 radicals and pulmonary vascular reactivity in rat lung. J. Appl. Physiol. 1989, 67, 1903–1911.
- Waypa, G.B.; Osborne, S.W.; Marks, J.D.; Berkelhamer, S.K.; Kondapalli, J.; Schumacker, P.T. Sirtuin 3 deficiency does not augment hypoxia-induced pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2013, 49, 885–891.
- Post, J.M.; Hume, J.R.; Archer, S.L.; Weir, E.K. Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. Am. J. Physiol. 1992, 262, C882–C890.
- Michelakis, E.D.; McMurtry, M.S.; Wu, X.C.; Dyck, J.R.; Moudgil, R.; Hopkins, T.A.; Lopaschuk, G.D.; Puttagunta, L.; Waite, R.; Archer, S.L. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: Role of increased expression and activity of voltage-gated potassium channels. Circulation 2002, 105, 244–250.
- Hong, Z.; Kutty, S.; Toth, P.T.; Marsboom, G.; Hammel, J.M.; Chamberlain, C.; Ryan, J.J.; Zhang, H.J.; Sharp, W.W.; Morrow, E.; et al. Role of dynamin-related protein 1 (Drp1)-mediated mitochondrial fission in oxygen sensing and constriction of the ductus arteriosus. Circ. Res. 2013, 112, 802–815.
- Maggiorini, M. Prevention and treatment of high-altitude pulmonary edema. Prog. Cardiovasc. Dis. 2010, 52, 500–506.
- Reeve, H.L.; Michelakis, E.; Nelson, D.P.; Weir, E.K.; Archer, S.L. Alterations in a redox oxygen sensing mechanism in chronic hypoxia. J. Appl. Physiol. 2001, 90, 2249–2256.
- Archer, S.L.; Gomberg-Maitland, M.; Maitland, M.L.; Rich, S.; Garcia, J.G.; Weir, E.K. Mitochondrial metabolism, redox signaling, and fusion: A mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H570–H578.
- Wolin, M.S. Novel role for the regulation of mitochondrial fission by hypoxia inducible factor-1α in the control of smooth muscle remodeling and progression of pulmonary hypertension. Circ. Res. 2012, 110, 1395–1397.
- Marsboom, G.; Toth, P.T.; Ryan, J.J.; Hong, Z.; Wu, X.; Fang, Y.H.; Thenappan, T.; Piao, L.; Zhang, H.J.; Pogoriler, J.; et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ. Res. 2012, 110, 1484–1497.
- Hickey, M.M.; Richardson, T.; Wang, T.; Mosqueira, M.; Arguiri, E.; Yu, H.; Yu, Q.C.; Solomides, C.C.; Morrisey, E.E.; Khurana, T.S.; et al. The von Hippel–Lindau Chuvash mutation promotes pulmonary hypertension and fibrosis in mice. J. Clin. Investig. 2010, 120, 827–839.
- Ang, S.O.; Chen, H.; Hirota, K.; Gordeuk, V.R.; Jelinek, J.; Guan, Y.; Liu, E.; Sergueeva, A.I.; Miasnikova, G.Y.; Mole, D.; et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat. Genet. 2002, 32, 614–621.
- Bonnet, S.; Michelakis, E.D.; Porter, C.J.; Andrade-Navarro, M.A.; Thébaud, B.; Haromy, A.; Harry, G.; Moudgil, R.; McMurtry, M.S.; Weir, E.K.; et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation 2006, 113, 2630–2641.
- Piao, L.; Sidhu, V.K.; Fang, Y.H.; Ryan, J.J.; Parikh, K.S.; Hong, Z.; Toth, P.T.; Morrow, E.; Kutty, S.; Lopaschuk, G.D.; et al. FOXO1-mediated upregulation of pyruvate dehydrogenase kinase-4 (PDK4) decreases glucose oxidation and impairs right ventricular function in pulmonary hypertension: Therapeutic benefits of dichloroacetate. J. Mol. Med. 2013, 91, 333–346.
- Fang, Y.H.; Piao, L.; Hong, Z.; Toth, P.T.; Marsboom, G.; Bache-Wiig, P.; Rehman, J.; Archer, S.L. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: Exploiting Randle’s cycle. J. Mol. Med. 2012, 90, 31–43.
- Ryan, J.J.; Marsboom, G.; Fang, Y.H.; Toth, P.T.; Morrow, E.; Luo, N.; Piao, L.; Hong, Z.; Ericson, K.; Zhang, H.J.; et al. PGC1α-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2013, 187, 865–878.
This entry is offline, you can click here to edit this entry!