Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Amyotrophic Lateral Sclerosis (ALS) is a debilitating neurodegenerative condition characterized by the progressive degeneration of motor neurons. Despite extensive research in various model animals, the cellular signal mechanisms of ALS remain elusive, impeding the development of efficacious treatments. Among these models, a well-characterized and diminutive organism, Caenorhabditis elegans (C. elegans), has emerged as a potent tool for investigating the molecular and cellular dimensions of ALS pathogenesis.
- ALS
- cellular mechanism
- therapeutic application
1. Brief Introduction of ALS
Motor Neuron Disease (MND) constitutes a group of disorders, including, but not limited to, Amyotrophic Lateral Sclerosis (ALS), Progressive Spinal Muscular Atrophy, Primary Lateral Sclerosis, and Progressive Bulbar Palsy. These disorders share a common feature: damage to upper and lower motor neurons, resulting in the loss of essential motor function [1,2]. Typically, individuals diagnosed with MND manifest symptoms such as muscle wasting and limb weakness. In addition to motor impairments, they may also encounter challenges related to language and swallowing [3]. As MND progresses, most patients succumb to complications such as pneumonia or respiratory failure [4]. This review specifically addresses the intricate mechanisms underlying ALS, recognized as the most prevalent adult-onset neurodegenerative form of MND. We predominantly summarize the molecular and cellular pathways from studies conducted with the small model animal Caenorhabditis elegans (C. elegans).
ALS, commonly known as Charcot’s disease or Lou Gehrig’s disease, is a motor neuron disease (MND) affecting both upper and lower motor neurons within the central nervous system, governing voluntary muscle movement. Clinically, ALS is characterized by muscle rigidity and the gradual weakening of limbs and bulbar muscles, leading to varying degrees of difficulty in speech, swallowing, and respiration [4,5]. It is noteworthy that functions such as bladder control, bowel movements, and eye movements typically remain unaffected until the advanced stages of the disease [6]. In addition to muscle dysfunction, 30–50% of ALS patients present with cognitive and other nervous system deficits. Common cognitive symptoms in ALS patients include challenges in social cognition, verbal memory, language, and executive function [7]. Approximately 15% of cases with observed cognitive impairment exhibit visible atrophy in the frontal and/or temporal lobes, resulting in behavioral changes or language impairment meeting the criteria for frontotemporal dementia (FTD) [8]. These observations indicate the complexity of ALS as a progressive disease involving functional deficits in multiple tissues.
2. Pathogenic Mechanisms of ALS Implicated in C. elegans
- (1)
-
Innate immunity
Data from clinical studies show that multiple genetic mutations linked to ALS enhance neuroinflammation, which provides compelling evidence for immune dysregulation in the pathogenesis of ALS [30,79,80]. Although C. elegans lacks a classical inflammatory response or inflammatory cytokines analogous to mammals, it possesses an innate immune system. Notably, mutated ALS-associated proteins have been found to activate an innate immune response in C. elegans [81,82,83]. In C. elegans strains expressing mutant TDP-43 or FUS in their motor neurons, age-dependent motility defects culminate in paralysis and motor neuron degeneration at a rate significantly higher than that observed in wild-type TDP-43 or FUS control strains. By examining the expression of immune response proteins, including NLP-29 (an antimicrobial, neuropeptide-like protein expressed in hypodermal and intestinal tissue), it is evident that the expression of mutant TDP-43A315T or FUSS57∆ protein triggers the upregulation of immune response genes, suggesting that innate immune response may contribute to motor neuron neurodegeneration. Furthermore, mutated ALS-associated proteins trigger an TIR-1/Sarm1 immune pathway innate immune response in C. elegans motor neurons [84,85]. Loss-of-function mutations in tir-1, associated downstream kinases, and the transcription factor atf-7 collectively serve to suppress motor neuron degeneration, further supporting the notion that the innate immune system is involved in ALS models.
Despite knowing the importance of the immune response in ALS, there are many details that have not yet been elucidated. For instance, mutated ALS-associated proteins in neurons may elicit an immune response as part of a host defense reaction against pathogens or aid tissue repair. The molecules necessary to induce the expression of NLP-29 also need to be further explored. Chikka et al. reported that activation of the mitochondrial p38MAPK/ATF-7 immune pathway in the intestine is neuroprotective and sufficient to prevent rotenone-induced neurodegeneration [86]. Due to mitochondrial dysfunction being a prevalent feature of many neurodegenerative diseases, including ALS [87], the mitochondria-regulated immune pathway may also be involved in C. elegans motor neuron degeneration. C. elegans’ innate immune response coordinates its activity with the insulin/IGF-1 pathway [88], suggesting that insulin-related immune pathways are also worth investigating. Nevertheless, these studies reveal that cell-based strategies that enhance anti-inflammatory reactivity and reverse immune dysregulation offer the potential to slow disease progression and improve the quality of life of patients with ALS.
- (2)
-
Autophagy
It is widely acknowledged that the dysregulation of autophagy in motor neurons is a pivotal event in ALS [89,90,91]. Particularly, intensified immunoreactivity in the cytoplasm of motor neurons for microtubule-associated protein 1 light chain 3 (LC3), which is a marker of autophagosome, is frequently observed in the spinal motor neurons of ALS patients [92,93]. Consistent with this observation, C. elegans with dynactin 1 knockdown (dnc-1(RNAi) worms) in ventral motor neurons under the control of the Pacr-2 promoter exhibited notable motor impairments, coupled with axonal and neuronal degeneration. Notably, the autophagosomes were easily trapped where the axon was tight, curved, or at spheroids. The phenomenon was followed by the accumulation of autophagosomes distal to the trapped sites [60]. Given that autophagosomes serve as cargo for dynein/dynactin complexes and play a pivotal role in the turnover of various organelles and proteins, the accumulation of autophagosomes suggests a potential contribution of dysfunctional autophagy to motor neuron degeneration in ALS. Indeed, the introduction of pharmacological disruptions to autophagy, using 3-MA, resulted in locomotory defects and axonal degeneration mirroring those observed in dnc-1(RNAi) worms. This implies that a compromised autophagy system alone is adequate to induce motor neuronal degeneration [60].
The contribution of defective autophagy to neuronal dysfunction in ALS is well-documented by autophagy-related genes [30,94,95,96,97,98]. The selectivity of autophagy is mediated by autophagy receptors that recognize and deliver cargoes to autophagosomes for degradation. SQSTM1/p62 is an autophagy receptor that is commonly found in protein aggregates in ALS brains. Related reports showed that SQSTM1 promotes the clearance of stress granules, a hallmark of ALS, via selective autophagy [99,100]. The main autophagy process is proteotoxic stress, which activates serine/threonine kinase TBK1, promotes phosphorylation of autophagy receptor SQSTM1, and activates selective autophagy. In contrast, ALS-linked mutations of TBK1 or SQSTM1 reduce SQSTM1 phosphorylation and compromise ubiquitinated cargo binding and clearance (Figure 1). The accumulation of SQSTM1 implicates a disturbance of the selective autophagy pathway [101]. Corresponding with the accumulation of autophagosomes, SQSTM1/p62 has been observed to accumulate in the motor neurons of ALS patients [102]. This observation aligns with findings demonstrating elevated levels of LC3-positive autophagy vesicles in the motor neurons of ALS patients with FUS mutations [103]. Notably, in an ALS C. elegans model involving overexpressing human mutant FUS proteins, a gain of toxic function mechanism disrupts basal neuronal autophagy. There was an increased accumulation of SQST-1 that disrupts neuromuscular function in stress conditions, the C. elegans ortholog for SQSTM1/p62, in motor neurons [38], reinforcing the link between autophagy dysfunction and ALS pathology. Conversely, the loss of sqst-1 suppresses both neuromuscular and stress-induced locomotion defects in FUS-associated ALS worms. It is worth mentioning that this suppression likely does not accompany a correction of neuronal autophagy defects [38], indicating that SQST-1 operates through an autophagy-independent pathway or alternative mechanisms to ameliorate ALS-related locomotion impairments [104,105]. The mutation of a single autophagy receptor can induce the decline of autophagy and lead to abnormal protein accumulation. But when autophagy receptors are passively increased, reducing autophagy levels may have a positive effect. Thus, the treatment of ALS requires multi-factorial and systematic consideration.
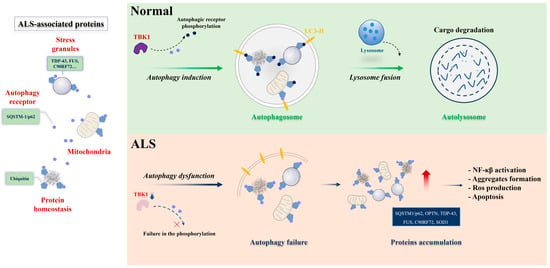
Figure 1. Selective autophagy under physiological and ALS pathological conditions. Protein aggregates, stress granules, and dysfunctional mitochondria serve as substrates for selective autophagy degradation. In physiological conditions (upper panel), these substrates are bound by selective autophagy receptors, such as SQSTM-1/p62 (represented in blue circle), via ubiquitin-binding domains (ubiquitin, in pentagon). The selective autophagy receptors associate with LC3-II proteins in the autophagosome (represented in yellow) or other members of the autophagy machinery. Posttranslational modifications in the receptors can enhance binding with ubiquitinated substrates or the LC3-II protein. TBK1 is one of the main kinases acting in this process. The cargo-receptor- LC3-II complexes are then sequestered by de novo double-membrane vesicles called the autophagosome, which fuses with the lysosome for the final degradation. Under ALS conditions (lower panel), failure in selective autophagy can occur through mutations in the genes encoding the receptors themselves or in the kinase, reducing the activity of the pathway and promoting the accumulation of toxic substrates for motor neurons. Figure was generated by PowerPoint 2013.
Furthermore, increased expression levels of autophagic genes by daf-2(e1370) have been shown to protect C. elegans motor neurons against the toxicity of human SOD-1(G93A) [41]. Metformin, the globally prescribed biguanide drug worldwide for the treatment of type II diabetes, alleviates motor dysfunction in human SOD-1(G93A)-associated ALS worms, partly through enhancement of autophagy [106]. Although not explicitly validated in C. elegans models, investigations in other model systems have demonstrated cross-regulation between TDP-43 pathology and autophagy [107,108]. These findings imply the existence of supplementary autophagy mechanisms in ALS [109]. In conclusion, enhancing autophagy emerges as a novel and significant therapeutic target for addressing motor neuron degeneration in ALS.
- (3)
-
Protein homeostasis
Protein homeostasis (proteostasis) is carefully maintained through a finely regulated and interconnected network of biological pathways, crucial for preventing the accumulation and aggregation of damaged or misfolded proteins [110]. Conversely, the breakdown of proteostasis has been implicated in the etiology of various neurodegenerative diseases, including ALS [111]. Zhang et al. conducted genetic analysis and expression profiling of loss-of-function tdp-1 mutants, elucidating the role of C. elegans TDP-1 (nematode TDP-43 ortholog) in regulating protein homeostasis. In diverse proteotoxicity models, the loss of TDP-1 alleviated protein aggregation and neuronal dysfunction. Their findings suggest that TDP-1 loss may modify global RNA levels, consequently impacting protein homeostasis and prompting cellular adaptation to stress on protein quality control systems [70]. Transgenic C. elegans models that express human TDP-43 variants displayed severe locomotor defects associated with the aggregation of TDP-43 in neurons [50]. Notably, the neurotoxicity and protein aggregation of TDP-43 were influenced by environmental temperature, and heat shock transcriptional factor 1 (HSF-1) played a role through protein quality control [112], indicating that a deficiency in protein quality control serves as a risk factor for TDP-43-associated ALS [50].
Aging-related neurodegeneration associated with TDP-43 is further connected to protein misfolding. The well-explored regulatory mechanism governing longevity and proteostasis involves the modulation of the insulin/IGF-1 signaling pathway through phosphorylation [113,114]. In the nematode C. elegans, the downstream receptor of insulin molecule, daf-2, has demonstrated the capability to counteract the shortened lifespan resulting from FUS overexpression [50,115]. These findings illuminate the intricate network of cellular mechanisms, notably the insulin/IGF-1 pathway, designed to preserve protein homeostasis in the presence of environmentally induced damage or genetically encoded misfolded proteins.
- (4)
-
Energy metabolism
While the intricate mechanisms underlying the association between mutant TDP-43, FUS, and Amyotrophic Lateral Sclerosis (ALS) are intricate and multifaceted, an accumulating body of evidence supports the presence of dysregulated energy metabolism in both ALS patients and relevant models [116]. The AMP-activated protein kinase (AMPK) serves as a pivotal cellular energy sensor. Upon activation, AMPK restores energy homeostasis by facilitating catabolic pathways, thereby promoting ATP generation [117]. Notably, heightened AMPK activation has been documented in the motor neurons of ALS patients, displaying a notable correlation with the extent of cytoplasmic mislocalization of TDP-43 [118]. These observations establish a clear link between energy depletion in human motor neurons and the pathological presence of TDP-43 in ALS. In line with this correlation, reducing AMPK activity has been demonstrated to ameliorate disease outcomes both in vitro and in C. elegans models expressing mutant SOD1 or TDP-43 [45]. Although a definitive mechanistic link between AMPK-regulated energy metabolism and TDP-43 mislocalization remains elusive, a proposed hypothesis suggests that the aggregation of TDP-43 stems from AMPK-mediated inhibition of nucleocytoplasmic transport.
Mitochondrial dysfunction is a prevalent characteristic of ALS [119,120]. Mutant forms of TDP-43, SOD-1, and FUS proteins have been implicated in disrupting mitochondrial structure and function [121,122,123]. This dysfunction induces an energy imbalance within neurons, affecting energy production and utilization. Specifically, FUS mutations have been linked to disturbances in mitochondrial function, potentially impeding the neurons’ capacity to generate ATP [121]. Consequently, compromised energy metabolism may play a role in the vulnerability and degeneration of motor neurons in ALS. Enhancing mitochondrial biogenesis emerges as an appealing therapeutic strategy for ALS. It is important to note that, despite the absence of current evidence from the C. elegans model, gaining further insights into these altered physiological processes in neurons—particularly by expressing mutant TDP-43 or FUS in C. elegans—becomes crucial for a more comprehensive understanding of ALS pathogenesis, specifically pertaining to energy metabolism.
In summary, mitochondria play a key role in ATP supply to cells via oxidative phosphorylation. Decreased ATP levels emerge as a common feature in ALS. It is conceivable that, in line with the high energy demands of neurons, gradual depletion of ATP, due to reduced respiration, may trigger neuronal degeneration [87,116]. In addition, the mitochondrial REDOX reaction is associated with the production of SOD. A lower concentration of ROS is essential for normal cellular signaling, whereas a higher concentration and long-term exposure of ROS cause damage to cellular macromolecules such as DNA, lipids, and proteins, ultimately resulting in necrosis and apoptotic cell death [124]. Altogether, these data suggest that bioenergetic abnormalities are likely to be pathophysiologically relevant to ALS disease.
This entry is adapted from the peer-reviewed paper 10.3390/cells13010099
This entry is offline, you can click here to edit this entry!