Candidiasis is a highly pervasive infection posing major health risks, especially for immunocompromised populations. Pathogenic Candida species have evolved intrinsic and acquired resistance to a variety of antifungal medications.
1. Introduction
1.1. Candidiasis
Candidiasis is an infection caused by the overgrowth of pathogenic yeast from the
Candida genus [
1]. Its prevalence accounts for the most common type of opportunistic fungal infection affecting human heath globally, with more than a billion cases on a yearly basis [
2,
3]. Typically, yeast can live harmlessly on the host’s mucosal tissues, such as in the oral cavity, gastrointestinal mucosa and vaginal mucosa, unless balance is disrupted [
4,
5]. The immunosuppressed, elderly population and palliative patients are highly susceptible to
Candida infections [
6]. Oral candidiasis results in local oral pain and discomfort, enhanced oral dryness, loss of taste and aversion to food and may lead to secondary complications [
7,
8]. Failure to treat candidemia in sufficient time is associated with a significant risk of mortality, especially in severe cases that have evolved into invasive fungal diseases (IFDs) [
9,
10].
Fungal resistance to traditional antifungal treatments has emerged as a significant and continued threat, yet it has received limited focus until recently in the fight against antimicrobial resistance [
3,
10]. Numerous factors contribute to the rising incidence and expanding geographic reach of pathogenic
Candida infections. These includes the increase in immunocompromised patients, fungi continuing to evolve resistance to treatments and the limited access to timely diagnostic options for clinicians [
10,
11]. Furthermore, these fungal species grow more optimally at higher temperatures. As a result, global warming enhances the growing threat of fungal infection spread increasing beyond the load that health care can manage [
11]. The worldwide impacts of pathogenic
Candida and resistant infections include the increased burden on the healthcare system, higher costs and fatalities arising from treatment failures [
10,
12]. Knowledge of the molecular mechanisms underlying antifungal resistance is important to help drive the development of novel fungal therapeutics and diagnostics.
1.1.1. Candida Species of Interest
Phylogenetic categorization of
Candida yeast suggests polyphyly and pathogenic diversity among members [
18]. Three of these species (
C. albicans,
C. parapsilosis and
C. tropicalis) belong to the CTG clade, which contains most pathogenic
Candida species, while
P. kudriavzevii is more closely related to a wine-making yeast (
Brettanomyces bruxellensis) [
18]. Members of the CTG clade have a divergence in their genetic code compared to other Saccharomycotina subphylum yeast [
19,
20]. These species are categorized based on the CTG codon being transcribed and translated into serine instead of a typical leucine [
19,
20].
C. glabrata is part of the
Nakaseomyces clade and was recently renamed
Nakaseomyces glabrata for improved classification [
21]. Despite this species being one of the few pathogenic members of its clade, it is the second most common cause of candidiasis globally [
18]. The consequences of genetic alterations in
N. glabrata may diverge from typical
Candida species because it is a haploid organism [
22]. Resistance could arise at a higher rate because a single recessive point mutation can present phenotypically due to a haploid genome. This contrasts with other
Candida species that are diploid and therefore may require two copies of the mutated gene to present with resistance [
23].
1.1.2. Candida auris
Candida auris of the CTG clade is listed under critical priority in the FPPL due to its high infectiousness, global spread and high fatality risk [
17,
18,
24,
25]. Numerous species isolates have been identified as displaying resistance to several antifungals [
26,
27,
28]. An update on
C. auris released by Public Health Ontario (2023) indicated high rates of resistance to azole drug fluconazole (87–100%), while polyene amphotericin B and echinocandin resistances are cited less frequently, with ranges of 8–35% and 0–8%, respectively [
29]. The CDC reported a similar rate of approximately 30% for polyene-resistant strains [
30]. At least 4% of global cases of
C. auris infections display multidrug resistance to all three antifungal types, which can make adequate clinical treatment especially difficult [
29].
1.2. Primary/Intrinsic Resistance vs. Secondary/Acquired Resistance
Fungal resistance can be divided into primary/intrinsic and secondary/acquired resistance. Some fungal species are intrinsically resistant to a specific antifungal drug because of innate functional or structural attributes. This stable feature is seen in all strains from the same species and has not evolved due to previous antifungal exposure [
1]. One example is the intrinsically fluconazole-resistant
P. kudriavzevii [
32,
33]. Alternatively, acquired resistance can evolve in strains of a
Candida species that are typically susceptible to an antifungals. This secondary form of resistance usually develops after prolonged treatment in a clinical or in vitro setting [
34]. Mutations or chromosomal rearrangements can cause an overexpression of genes that override the effects of antifungal activity or the fungal stress response [
34]. This change can revert to the original state once the pressure of the drug treatment is reduced or removed. Some mutants may retain the resistant phenotype regardless of future drug pressure [
34].
Antifungal action can be evaded by pathogenic yeast by two main methods: (1)
An alteration of the interaction between drug and target. This can result from either a change in the target protein amino acid (aa) sequence and, consequently, its structure or target protein overexpression. Alternatively, (2)
the cytoplasmic drug concentration can be reduced via cell wall modifications that decrease drug absorption into the cell or the overexpression of efflux pumps that promote the export of drug molecules out of the cell [
34].
Multidrug resistance (MDR) occurs when these mutations accumulate in the yeast genome in target pathways, which limits the amount of treatment options available [
23,
35]. An example of MDR was observed in
C. albicans isolates with resistance to both fluconazole and clotrimazole [
36]. Detecting mutant genotypes with acquired resistance in a timely manner could be an independent and useful predictive risk factor for treatment failure [
34,
37,
38].
1.3. Standardized Measures of Susceptibility Testing
Minimum inhibitory concentrations (MICs) calculated with in vitro broth microdilution susceptibility testing are used to categorize dose-dependent resistance in
Candida species [
39,
40,
41]. There are two main standardization methods that outline the established breakpoint concentrations for ranges of resistance: those of the
Clinical Standards Laboratory Institute (
CLSI—North America) and the
European Committee on Antimicrobial Testing (
EUCAST) [
40,
41,
42]. Considering that
C. parapsilosis cryptic species
C. orthopsilosis and
C. metapsilosis are of low prevalence, the MIC breakpoint data for
C. parapsilosis can be applied in cases when further species characterization has not been completed [
43,
44,
45,
46,
47]. One trend identified in the data is the tendency of high-resistance concentrations in
N. glabrata for fluconazole, whereas lower MICs are implicated with the use of echinocandins for this species.
2. Antifungal Classes and Frequency of Resistance
A range of antifungal drug classes is available to target various molecules and pathways associated with pathogenic
Candida infections. The reviews by Bhattacharya et al. (2020) and Tilley and Tharmalingam (2022) provide an excellent summary of the four primary antifungal drug classes: azoles, polyenes, echinocandins and nucleoside analogs [
1,
55]. The section below summarizes the mechanisms of action for each antifungal drug class (
Figure 1), as well as relevant resistance profiles. The molecular structures of drugs from each antifungal class and key points for each type are presented in
Figure 2.
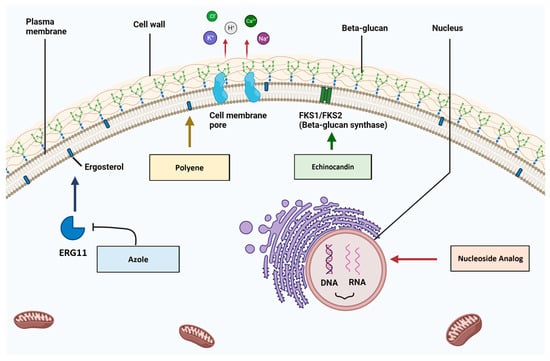
Figure 1. Mechanisms of antifungal action for the four main drug types. (1)
Azoles bind to and inhibit the Erg11 enzyme and subsequent ergosterol production. (2)
Polyenes bind to ergosterol and induce the formation of cell membrane pores, which cause intracellular ion leakage. (3)
Echinocandins bind to and inhibit beta-glucan synthase, which disrupts cell wall architecture. (4)
Nucleoside analogues are incorporated into nucleic acid molecules and disrupt DNA/RNA biosynthesis (created with
BioRender.com, accessed on 16 October 2023).
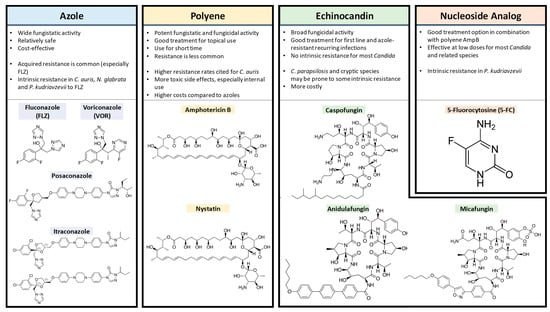
Figure 2. The key points for each of the four antifungal drug types with the chemical structures of members from each class. All drug structure images were obtained from
Wikimedia Commons.
2.1. Azoles
Azoles are five-membered heterocyclic compounds classified into two groups based on the number of azole-ring nitrogen atoms: imidazoles with two nitrogens, like clotrimazole, ketoconazole and miconazole; and triazoles with three nitrogens, like fluconazole, itraconazole and voriconazole [
55,
56]. This antifungal class inhibits the production of ergosterol, an important component of the fungal cell membrane. With wide fungistatic activity, it is a cost-effective and relatively safe treatment option [
57,
58]. Fungistatic effects result in the inhibition of yeast growth [
1]. Fluconazole has been prescribed as a first-line agent for fungal infections, and consequently, resistance has also been frequently cited [
59]. The development of second-generation triazoles like voriconazole, posaconazole and isavuconazole offers secondary options for resistant
Candida infections, although acquired resistance has been noted in past years [
1]. Another barrier to the successful treatment of candidiasis infections with azoles is varying pharmacokinetics. Some drugs of this type, like itraconazole, may have poor absorption. For internal use, the absorption can be improved with food intake [
60].
2.2. Polyenes
Polyene antifungals like amphotericin B and nystatin target the fungal plasma membrane by binding ergosterol molecules and forming pores that leak cell contents (monovalent ions K
+, Na
+, H
+ and Cl
−) [
67]. These are potent agents with fungicidal and fungistatic activity that are used in clinical practice for their effectiveness despite relatively higher rates of toxic side effects like kidney/liver issues or anaphylaxis [
50,
68,
69]. Fungicidal agents can kill infectious yeast cells directly [
1]. To limit the possibility of treatment toxicity, this class is best used for topical infections such as in the oral cavity and for a limited time course [
69]. Reports of isolated fungal strains with acquired polyene resistance are relatively rare, despite decades of use in the clinical setting [
70,
71]. This may be attributed to the effectiveness of their fungicidal activity in eliminating infections and thus preventing the evolution of stable resistant mutants.
2.3. Echinocandins
Echinocandins including caspofungin, micafungin and anidulafungin target the fungal cell wall, a feature not found in mammalian cells [
73]. Echinocandins are composed of cyclic hexapeptides with lipid side-chain modifications that enable antifungal action [
74]. They inhibit the synthesis of a major cell wall component via non-competitive binding to the Fks1 subunit of the β1–3 glucan synthase enzyme [
75]. This action promotes a fungicidal effect, as the cell wall integrity is compromised, with increased permeability and subsequent amino acid leakage [
74].
C. albicans tends to be the most susceptible to caspofungin, followed by
N. glabrata,
C. tropicalis,
P. kudriavzevii,
C. parapsilosis and
M. guilliermondii [
74]. The last two species listed seem to have more naturally arising FKS1 point mutations; thus,
C. parapsilosis and
M. guilliermondii appear to be more intrinsically echinocandin-resistant [
77,
78].
Secondary resistance to echinocandins has been observed and linked to point mutations in the FKS1 gene that alter antifungal binding capacity [
79]. Strain viability may be compromised as indicated by the reduced virulence seen in multiple echinocandin-resistant
Candida species [
66].
2.4. 5FC
5-Fluorocytosine (5FC) can be used to target and disrupt nucleic acid biosynthesis within the cell [
80]. This nucleoside analog used in conjunction with polyene amphotericin B is a reliable option for difficult-to-treat
Candida infections and cryptococcal meningitis [
81,
82,
83]. The minimum inhibitory concentration for 90% of fungal growth (MIC90) (National Committee for Clinical Laboratory Standards (NCCLS)) determined using the antifungal susceptibility testing method has been cited from 0.12 to 1 ug/mL depending on the species and sample [
83].
3. The Ergosterol Biosynthesis Pathway and Antifungal Resistance
Azoles and polyenes target the ergosterol biosynthesis pathway, specifically the 14α–demethylase enzyme (Erg11) and ergosterol molecules respectively (
Figure 3) [
1]. Sterols, along with sphingolipids, can form lipid rafts within the fungal cell membrane that contain proteins crucial for yeast survival, like stress response, signaling and nutrient transport proteins [
1]. There are 25 different pathway enzymes involved in the formation of ergosterol [
85]. Azoles primarily exhibit fungistatic action via non-competitive binding to the Erg11 enzymatic active site, which inhibits its activity and results in an overall decrease in cellular levels of ergosterol [
86].
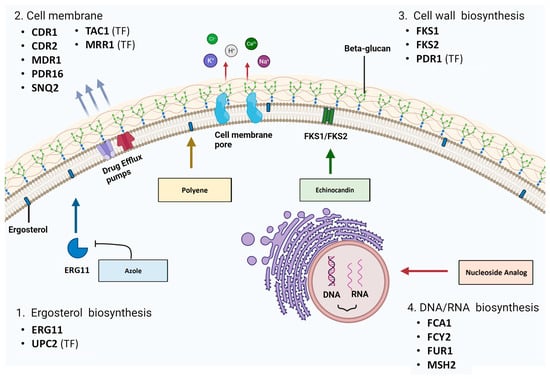
Figure 3. Genes associated with antifungal resistance in drug target pathways: (1) ergosterol biosynthesis, (2) cell membrane, (3) cell wall biosynthesis and (4) DNA/RNA biosynthesis (created with
BioRender.com, accessed on 16 October 2023).
3.1. ERG11
By 2010, over 160 amino acid substitutions had been identified in the ERG11 gene, each with varying genetic consequences [
87,
88,
89]. Many single substitutions are synonymous and have no impact on gene function. In addition, non-synonymous single-nucleotide changes occurring in
Candida strains do not inherently contribute to antifungal resistance. For example, White et al. (2002) identified D116E and E266D, the two most frequent ERG11 substitutions in one sample set, using restriction fragment length polymorphism (RFLP) analysis, with no consistent correlation [
36]. These instances indicate that there is natural genetic variation within each fungal species and that allelic polymorphisms are relatively common. Mutations present in both resistant and susceptible samples are an indicator that the alteration is not directly implicated in conferring antifungal resistance [
86].
Candida spp. have acquired azole resistance via ERG11 point mutations that typically lower azole binding affinity to the Erg11 active site. Additionally, gain-of-function mutations in upstream transcriptional regulators can increase ERG11 expression and confer resistance. [
34,
86]. Point mutations resulting in a defective Erg11 enzyme unable to bind to azoles have been clustered in three hotspot regions: 105–165, 266–287 and 405–488 [
94].
Polymorphisms can have different biological impacts, depending on whether they are singly present or in combination with other relevant SNPs. Resistance levels can be enhanced when some mutations with moderately low impact alone are present simultaneously with other resistance mutations [
86,
103,
104].
The variety of currently available azole medications have different structural features (e.g., short and long chains); therefore, mutations affecting their efficacy may differ. For example, ERG11 point mutations K128T and Y132H may affect the ability of fluconazole or voriconazole molecules to enter or bind the target active site. Mutations in other gene sequences can also confer resistance, such as the G464S mutation, which affects haem coordination due to its location near a key cysteine residue [
93]. These mutations do not have the same binding inefficiency for posaconazole and itraconazole treatments, suggesting that these two antifungals have other key interaction sites within the Erg11 protein [
93].
3.2. Mutations in Transcriptional Regulators
Zn2-Cys6 transcription factor
uptake control 2 (Upc2) regulates most of the genes in the ergosterol biosynthesis pathway on some level. Gene overexpression of UPC2 can be induced upon azole exposure and can sufficiently compensate for the inhibition of target enzymes. Gain-of-function (GOF) mutations in UPC2 can drive this gene overexpression and fluconazole resistance [
101]. For example, a series of studies of azole-resistant clinical isolates identified the A643V substitution in the UPC2 gene, which was validated in vitro to confer resistance [
101,
107]. This mutation and possibly others within this gene sequence region (G648D) may cause Upc2 to be released from a repressor, inducing hyperactivity [
107]. However, alternate models exist that involve the sterol regulator, SREBP [
107].
A genome-wide ChIP (chromatin immunoprecipitation) study used to identify Upc2-bound gene promoters identified up to 202 genes, including UPC2 itself [
111]. Other upregulated genes were found to be involved in ergosterol biosynthesis; oxidoreductase activity; and numerous drug efflux pumps, including MDR1 (MFS-transporter) and CDR1 (ABC-transporter) [
101].
3.3. Other ERG Genes and Toxic Diol Formation
The inhibition of Erg11 alters pathway products and induces the synthesis of a fungistatic toxic diol (14α-methylergosta 8–24 (28) dienol) by downstream enzymes (Erg3, Erg6, Erg25, Erg26 and Erg27) [
1]. Erg3 is a C5 sterol desaturase enzyme needed for the conversion of episterol to ergostatrienol [
112]. When its expression is inhibited by mutation or deletion, the reduction in toxicity due to the inhibition of toxic diol formation is sufficient to confer resistance in some
Candida spp. [
113,
114,
115]. However, ERG3 inactivation appeared to minimally contribute to azole resistance in a wide range of studied clinical
C. albicans [
116]. Q139A substitution in ERG3 has been identified from
N. glabrata clinical isolates with azole resistance [
99].
Deletion of ERG pathway genes such as the ERG6 gene can impact resistance to other antifungals. This ∆24 sterol C-methyl transferase is non-essential for ergosterol biosynthesis but it is needed for toxic diol formation. Its disruption contributes to azole resistance in
C. albicans [
117,
118].
One differential gene expression analysis of a lab-generated
C. albicans strain with resistance to fluconazole and amphotericin B identified numerous upregulated ergosterol pathway genes, including ERG5, ERG6 and ERG25 [
112]. Additionally, genes involved in cell stress responses were found to be upregulated, including DDR48 and RTA2 [
112]. In
C. albicans, RTA2 is a key gene in calcium signaling pathways, and it has been shown to modulate azole resistance, including biofilms [
120].
4. Cell Membrane Proteins and Antifungal Resistance
Two types of membrane transporters have been implicated in azole resistance: ABC-Ts (ATP-binding cassette transporters) and MFS-Transporters (major facilitator superfamily transporters) [
36,
110,
121,
122]. ABC-Ts facilitate the movement of molecules across membranes using energy derived from ATP hydrolysis, while MFS-Ts require a proton gradient across the plasma membrane to transport foreign molecules out of the cell [
1]. Both types of transporters can bind azoles as a substrate, and the drug can be exported out of the cell. This decreases the intracellular drug concentration and allows cells to circumvent the antifungal effects [
1]. An additional MLT1 (ABC-T) transporter has been implicated in
C. albicans resistance. This multidrug resistance protein (MRP) is localized to the vacuolar membrane and can import azole molecules into the vacuole for sequestration. Mutations in the MLT1 sequence can cause incorrect localization or the inability to bind and transport azoles [
123].
4.1. Drug Efflux Pump/Transporter Genes and Resistant Mutations
Despite the large number of transporters found in the
C. albicans genome, evidence of transporter overexpression in resistant clinical isolates is currently limited to ABC-Ts CDR1 and CDR2 and MFS-Ts MDR1 and PDR16 [
1,
111,
131,
133]. CDR1 and CDR2 overexpression is frequently observed in clinical isolates, and coregulation of these two pumps is evident [
36]. In fact, prolonged exposure to azoles can result in trisomy development of chromosome 3, which encodes CDR1 and CDR2, as well as subsequent overexpression of these genes [
124]. For azole-resistant
N. glabrata clinical isolates, overexpression of ABC-T genes CDR1, PDH1 (CDR2 in
C. albicans) and SNQ2 and MFS-T genes QDR2, FLR1 and PDR16 has been cited [
62,
104,
134,
135,
136,
137,
138,
139].
4.2. Transcriptional Regulators of Transporter Genes
Efflux pump overexpression can be further induced by upstream GOF mutations, regulating transcription factor genes [
59,
125,
126,
127,
128,
129,
130,
144,
145]. In
C. albicans, the CDR1, CDR2 and PDR16 transporters, as well as MDR1, are regulated by zinc-cluster (Zn2-Cys6) transcription factors Tac1 and Mrr1, respectively [
111,
121,
131,
133,
144,
146]. Other potential transcriptional regulators of CDR1 expression include Tup1 (thymidine uptake 1) and Ncb2 (β subunit of the NC2 complex) [
147]. For MDR1, additional transcriptional regulators are Cap1 (bZIP transcription factor) and Mcm1, but no antifungal-resistant mutations have been identified to date [
148,
149].
4.3. Post-Translational Regulation of Transporter Genes
In addition to transcriptional and translational control of efflux pump genes, there is evidence of post-translational regulation. For instance, mitochondrial biogenesis gene FZO1 is important for directing Cdr1 to the correct membrane [
153]. In FZO1 deletion mutants, Cdr1 was found to be mis-sorted to the vacuole, which was correlated with increased azole susceptibility [
153].
5. The Cell Wall Biosynthesis Pathway and Antifungal Resistance
5.1. FKS1 and FKS2 Sequence Mutations
Echinocandin resistance is associated with modifications to the FKS1 or FKS2 gene sequences, which code for the β1–3 glucan synthase enzyme [
156]. FKS1 mutations have been identified in resistant
C. albicans,
C. tropicalis,
P. kudriavzevii and
N. glabrata, while FKS2 mutations have only been identified in
N. glabrata [
157,
158,
159,
160]. No definitive intrinsic resistance has been established in any
Candida species, but secondary resistance can be acquired in individual isolates through point mutations. Notably,
C. parapsilosis and
Meyerozyma guilliermondii have a higher rate of spontaneously occurring FKS1 point mutations and may be considered more intrinsically resistant [
77,
78].
Significant GOF mutations in the FKS1 gene sequence in clinical isolates have been described, particularly in the hotspot regions of 637–654 and 1345–1365 [
74,
157,
162,
163,
164,
165,
166,
167,
168]. Walker et al. (2010) noted that non-synonymous substitutions at aa position 645 have been commonly observed. Here, serine substitutions with phenylalanine, proline or tyrosine have been cited [
163,
164,
169]. Hotspot mutations are usually dominant, and
C. albicans fungal cells only require one mutant allele for resistance to be conferred across the three echinocandins [
74].
Evidence of FKS2-resistant mutations were previously limited to in vitro experiments, but recently, such mutations have been identified in clinical
N. glabrata isolates [
34,
35,
173]. This coincides with evidence suggesting that FKS2 has higher levels of expression in
N. glabrata than FKS1 and that mutations may have a greater influence on echinocandin resistance in this species [
158].
5.2. Transcriptional Regulators of fks Genes
Transcriptional regulators upstream of
fks genes can also affect echinocandin resistance. In particular, point mutations in transcription factor PDR1 have been found in numerous resistant
N. glabrata isolates (
Figure 3).
5.3. Protein Analysis Associated with Echinocandin Resistance
Cell wall remodeling enzymes upregulated in previous protein studies include glucanosyl transferases Phr1, Phr2 and Crh, as well as chitin-glucanosyl transferase family proteins [
178,
179]. Proteomic analysis conducted using mass spectrometry (LC-MS/MS) revealed that levels of cell wall organization and maintenance proteins can differ between drug-resistant and susceptible strains in response to caspofungin treatment [
180,
181]. Differentially expressed enzymes related to cell wall synthesis and remodeling include Sun41, Gsc1, Pmt1, Mnt1, Als3, Als4, Ecm33 and Pga31 [
181]. Validated caspofungin tolerance regulators Cas5, Mkc1, Swi4, Gin4, Stt4, Ahr1 and Pkc1 were detected in an alternate screen [
182]. Furthermore, metabolic enzymes with immunogenic activity including Eno1, Fba1, Gpm1 and Pgk1 have also been observed to be released in
Candida cells exposed to caspofungin [
181,
183]. Finally, the Hsp90 molecule may have a regulatory role with key resistance regulators like Mkc1 from the Pkc1 signaling pathway [
184,
185]. These protein subsets may be good
Candidates for diagnostic markers that predict echinocandin resistance in
Candida species. Further validation across a wide range of antifungal-resistant clinical isolates is still needed [
181].
6. The Nucleic Acid Biosynthesis Pathway and Antifungal Resistance
The biosynthesis of nucleic acids (DNA and RNA) and subsequent protein synthesis in pathogenic fungi can be targeted with nucleoside analogue 5-fluorocytosine (5-FC). As a prodrug, it requires activation within the fungal cell via metabolism by the pyrimidine salvage pathway [83]. Then, it is incorporated as a toxic substrate, and the affected nucleotides have damaging effects on cell viability [83]. Membrane permeases encoded by FCY2 (cytosine permease) and other homologs (FCY21 and FCY22) are responsible for the active transport of 5-FC into the cell (Figure 3) [83]. 5-FC is then converted to toxic 5-fluoro-uridylate by enzymes encoded by fcy1 (cytosine deaminase) and FUR1 (uracil phosphoribosyltransferase (UPRT)) [83]. The FCY1 homologue in C. albicans and other Candida species is the FCA1 gene [189,190]. The lack of cytosine deaminase in mammalian cells prevents 5-FC conversion and subsequent toxic effects [191].
Resistance to 5-FC could arise with mutation or loss of any of the three key enzymes (FCY1, FCY2 or FUR1), as discovered in model organism yeast Saccharomyces cerevisiae [192,193]. Increased pyrimidine production in the fungal cell can also serve to circumvent toxic antifungal activity [82,83]. Kern et al. (1991) were among the first to identify the correlation between a point mutation (Arg134Ser) in the FUR1 gene and 5-FC resistance in S. cerevisiae yeast cells [194].
7. Biofilm Formation and Antifungal Resistance
7.1. Biofilm Formation during Antifungal Treatment
Over the course of antifungal treatment, a biofilm can develop and enable yeast cells to become more resistant [
209,
215]. Numerous classes of antifungals, including polyenes and azoles, have been cited as less effective over time, even within 72 h of biofilm development/maturation [
209]. For fluconazole treatment, resistance in
C. albicans with biofilms can be increased by up to 1000-fold compared to planktonic cells [
215,
217].
Multiple features of biofilms enable antifungal resistance aside from genetic mutations that typically drive resistance in planktonic cells, which adds to the complexity of treating this type of infection [
219]. These include physical protection, increased cell density of the microbe community, persister cells and extracellular vesicular secretion [
208,
219,
220].
7.2. The Roles of β-1,3 Glucan and Biofilm-Associated Antifungal Resistance
One of the main ECM components is β-1,3 glucan, which is synthesized by echinocandin target gene FKS1 [
223]. The polysaccharide molecules are primarily responsible for the sequestration of antifungal molecules, making them a prime target for treatment of
Candida infections with biofilms [
224]. Supplemental treatment with the β-1,3 glucanase enzyme can break down this molecule and subsequently disrupt the biofilm architecture [
225,
226].
7.3. Relevant Antifungal Resistance Genes in Biofilm-Associated Candida Infections
Candida strains associated with biofilm formation and resistance to antifungal treatment may have similar key genes implicated in planktonic resistant isolates. Changes in the expression of ergosterol biosynthesis pathway genes and in biofilm membrane composition appear to be linked to subsequent azole and polyene resistance [
230,
231,
232,
233]. Differential gene expression of beta-glucan synthesis-associated genes SKN1 and KRE1 was observed in biofilm-associated-resistant
Candida exposed to amphotericin B, in agreement with previous studies [
218,
230]. These relevant genes may be highlighted in the search for a suitable resistance biomarker.
Drug efflux pumps CDR1, CDR2 and MDR1, which are associated with azole resistance, may also be upregulated in biofilm-associated
Candida strains [
235,
236,
237,
238,
239,
240]. Interestingly, neither polyenes nor echinocandins have been implicated as a substrate for drug efflux pumps in
Candida infections of this type. This suggests that echinocandins could be a preferable treatment choice over azoles [
236,
237,
241].
8. Summary
With antifungal resistance being a continued problem, there is a need for the development of quick and reliable molecular diagnostic tests that detect organisms with intrinsic and/or secondary resistance due to the genetic mechanisms [34]. Additionally, faster methods of species identification would be useful, given the differences in frequency and impact of antifungal resistance among Candida species. Currently, PCR-based methods using fungal cultures are still the first option for species identification and detection of antifungal resistance in individual strains [105,167]. The usefulness of real-time testing for resistance in Candida species to modify the treatment course has been well documented.
Resistance can be acquired through a dynamic combination of numerous point mutations and other genetic or transcriptional alterations. A large-scale comparison between matched fluconazole-resistant and -susceptible C. albicans clinical isolates using microarray analysis identified almost two hundred genes (n = 198) that were differentially expressed [248]. In resistant isolates, multidrug resistance and oxidative stress response genes, among others, were found to be upregulated compared to susceptible samples [248]. This highlights the fact that a dynamic response that leads to antifungal resistance and identification of reliable biomarkers or gene expression profiles that different strains have in common would be beneficial to improve future treatment decisions.
To develop a dependable point-of-care (POCT) diagnostic assay for antifungal resistance, reliable biomarkers in Candida species need to be established. It is important to note that different non-synonymous substitutions and other genetic alterations may result in similar genetic effects.
This entry is adapted from the peer-reviewed paper 10.3390/cells12222655