Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Phenomics, the complexity of microglia phenotypes and their related functions compels the continuous study of microglia in disease animal models to find druggable targets for neurodegenerative disorders. Activation of microglia was long considered detrimental for neuron survival, but more recently it has become apparent that the real scenario of microglia morphofunctional diversity is far more complex.
- Alzheimer’s disease
- Hippocampus CA1
- Hippocampus CA3
- α7AChNR
- Aβ plaque
- rod microglia
1. Introduction
For over a century, the brain was seen as a network of neurons, dendrites, axons, and synapses in a space embedded by other cells which, like glue, fill the empty space. Nevertheless, proper recruitment, activation, and intercommunication among neurons and glia is of fundamental importance for the functional organization of the brain. The research is now focused on understanding how microglia engage in morphological, ultrastructural, transcriptional, proteomic, and epigenetic switches that influence their functions, their responses, and their effects on the surrounding cells, suggesting that microglia states are modulated by local cues (for a comprehensive review, see [1]).
While for most of the last century “activation” of microglia was considered solely detrimental for neuron survival, it was later demonstrated that microglia are fundamental in early brain development (for ref., see [2]), for synaptogenesis, synaptic maintenance, maturation and synaptic pruning (for ref., see [3]), and in normal learning and memory in mice [4,5,6,7].
Microglia (5–10% of brain cells in number) are myeloid cells that invade the brain early during development [8,9] and coordinate the interactions between the immune system and cognitive functions [3,10,11,12,13,14,15,16,17,18]. Early studies in adult mice demonstrate that microglia acquire diverse morphologies in different brain areas, from radially orientated arborized cells in the grey matter to longitudinally branched elongated cells in the white matter and compact amoeboid cells around the circumventricular organs [19]. For many years, ramified microglia (Figure 1A) were considered quiescent or ‘resting’, while they are now considered “homeostatic” [1].
In healthy conditions, microglia have small soma and fine, highly mobile ramified branches, which dynamically reorganize their shape and length, continuously elongating and withdrawing to patrol a defined, non-overlapping territory of the brain parenchyma [20] to detect and eliminate damaged neurons and maintain a healthy environment [21,22,23,24]. Recent studies have described hyper-ramified microglia (Figure 1D) in the medial prefrontal cortex of rats in response to chronic stress [25,26].
The ramified branches of microglia work as chemotactic sensors, moving their extensions towards injured cells for phagocytosis [27], and the impairment of their mobility can be deleterious in many conditions. Decreased migration of microglia hampers their phagocytic efficacy, increasing the degeneration of neurons and accumulation of toxic debris [28], and weakening microglia neuroprotective effects. Activation of microglia is a quick process that leads to morphological, phenotypic, and functional changes that stimulate the migration of microglia to the damaged brain area. Reactive microglia, after having fulfilled their phagocytotic function, regress rapidly to the homeostatic form [24]. Furthermore, during apoptotic clearance, spherical phagocytic pouches (ball) are formed at the tip of microglia terminal branches (chain), the so-called ball-and-chain structures (Figure 1C), which can phagocytose apoptotic debris [29] or a small quantity of other substances [29], with no modification of ramified morphology, in contrast to the phagocytosis performed by amoeboid microglia in pathological conditions (Figure 1E) [12]. In mouse organotypic hippocampal slices, ramified microglia exert neuroprotective effects during NMDA-induced excitotoxicity [30].
Microglia are plastic cells, and an oversimplified, already surpassed view, recognized two functional phenomic extremes acquired in response to cytokines, chemokines, and other soluble factors, namely the classical M1 pro-inflammatory and the M2 anti-inflammatory phenotypes [12,27,31,32,33,34,35]. M1 cells were classified as reactive cells that release pro-inflammatory cytokines, such as TNFα, IL-1, IL-6, and IL-18 [36], and have harmful properties. M2 were classified as non-reactive cells that secrete anti-inflammatory cytokines, such as IL-4, IL-10, IL-13, and TNF-ß, and have beneficial, neuroprotective properties, [36].
Nevertheless, the classification of microglia in these two all-or-none states [37] is too simplistic and does not correspond to the variety of different microglia phenotypes recently discovered [38,39]. Between these two extreme functional states, a plethora of phenotypically diverse intermediates with different functional states is now recognized. Indeed, a recent hypothesis based on accumulating data postulates that microglia, as neurons, exist physiologically as heterogeneous, mixed populations, which differ in their transcriptomic and morphofunctional characteristics. Consequently, microglia may exist in n possible phenomic states, diverse in health and disease conditions, and which depend not only on the type and intensity of insult and on the progression of the disease, but also on the brain structure where microglia are located [37,40,41,42,43,44]. The vast array of receptors expressed by microglia constantly surveying their surroundings constitutes a ‘sensome’, allowing detection and response to different stimuli that derive from sensory and behavioral experiences [1,45,46].
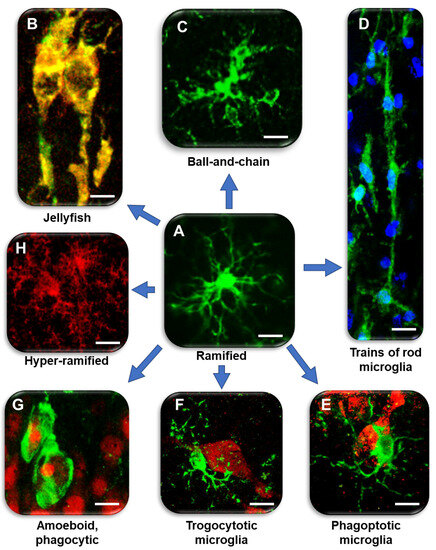
Figure 1. Diversity of microglia morphologies in the hippocampus. (A) Ramified, homeostatic microglia. IBA1: green (modified from [47]). Bar: 5 µm. (B) Jellyfish microglia in the ischemic CA1. IBA1: green, major histocompatibility complex type II (MHC II): red; merge: yellow–orange (modified from [48]). Bar: 2.5 µm. (C) Ball-and-chain-like structures at the end of microglia branches that can phagocytose small amounts of material. IBA1: green (modified from [49]). Bar: 5 µm. (D) Train of rod microglia in response to ischemia. IBA1: green; nuclei: blue (modified from [48]). Bar: 10 µm. (E) Phagoptotic microglia. IBA1: green; NeuN: red (modified from [50]). Bar: 5 µm. (F) Trogocytotic microglia. IBA1: green; NeuN: red (modified from [50]). Bar: 5 µm. (G) Amoeboid microglia with rounded morphology and phagocytosing pyknotic neurons after ischemia. IBA1: green; NeuN: red (modified from [48]). Bar: 10 µm. (H) Hyper-ramified microglia in CA1 of TGCRND8 mice. CD68: red (modified from [49]). Bar: 5 µm.
Microglia are anything but static, and, since they are exceptionally responsive to alterations in the surrounding environment, the states of microglia vary as a continuum rather than an all-or-none phenomenon. It will be interesting to understand whether microglia located in different brain areas acquire identical phenomics [51,52], or whether they react differently to the same insult. For instance, in the CA1 and CA3 hippocampus, microglia reactivity states are different in aging and acute inflammation [47,53], or after an excitotoxic insult [30]. Indeed, Vinet and colleagues [30] demonstrated that ramified microglia exert neuroprotective roles in pathologic processes, and that this function is region-specific, at least in the hippocampal areas. Region-specific differences in lysosome content and membrane properties of microglia [38,54], as well as the expression of genes related to the phagocytic capacity of microglia, have recently been demonstrated [55]. Furthermore, using scRNA-seq, it has been demonstrated that microglia populations display higher transcriptomic diversity in the developing, aged, and diseased brain [56,57] than in the adult brain. Many different receptors with a variety of functions are expressed by microglia. Their expression, and the outcome of their activation, depend not only on the pathological conditions, but also on the functional state of the cell. Depending on the nature of the ligand and on the receptor, downstream intracellular pathways translate their activation with detrimental or beneficial effects (for references, see [58]). The dysfunction of microglia has been described in many CNS disorders, such as AD [59], frontotemporal dementia [60,61], and PD [62].
2. Microglia Phenomics in Alzheimer’s Disease
Data from animal models of AD show that microglia are recruited at the site of Aβ deposition, and regulate Aβ levels in the brain [49], contributing to Aβ clearance and removal of cytotoxic debris from the brain [27,170,171,172,173]. Plaque-associated microglia inhibit additional fibrillization of Aβ and plaque growth [174], thus, protecting neighboring neurons [175]. However, the phagocytic activity and clearance capacity of microglia inversely correlate with Aβ plaque deposition and aging [105].
Microglia responses in AD are influenced by APOE and TREM2 [117]. TREM2 regulates microglia energetic and biosynthetic metabolism [176], maintaining the high activity microglia need to dispose of excess Aβ. However, the intense TREM2-dependent activation of microglia may in turn cause a harmful chronic inflammatory response of TREM2 [117]. Sustained activation of microglia can also cause phagoptosis of healthy neurons [171,177,178,179] intensifying neurodegeneration [65,180], and can cause phagocytosis of synapses in response to soluble Aβ [181], while microglia depletion prevents loss of neurons and dendritic spines [182], further suggesting a pathogenic role for microglia hyperactivation in AD. Variants of TREM2 impair microglia activation, phagocytic properties, inflammatory responses, energy metabolism, and plaque compaction, affecting the progression of AD [176,183,184]. Toll-like receptor 4 (TLR4) in microglia plays an important role in neuroinflammation [185], but studies on TLR4- and on TREM2-deficient mice give conflicting results on AD pathology [186]. In TREM2 deficient mice, loss of microglia clustering around Aβ plaques increases AD risk, supporting the idea that microglia are protective [175]. Nevertheless, it has also been shown that microglia associated with Aβ plaques have a neurodegenerative phenomic, regulated by the TREM2-APOE pathway, which suppresses the phagocytosis of apoptotic neurons [117]. TLR4, stimulated by both fibrillar and oligomeric forms of Aβ [187], seems to be protective [188]. Further, stimulation of Toll-like receptor 2 (TLR2) by fibrillar Aβ activates microglia into a more pro-inflammatory profile, with detrimental effects on AD pathology [187].
The age-related impairment of chemotactic sensors may weaken the neuroprotective activity of microglia which phagocytose Aβ fibrils less efficiently in old than in young mice [66]. On the contrary, amplified, exaggerated, or chronic microglia activation can lead to robust pathological changes and neurobehavioral complications, such as in chronic inflammatory diseases [39,98].
In both AD transgenic mice and human AD brains, a unique subtype of protective microglia, named disease-associated microglia (DAM), has recently been found [39]. DAM contribute to disease mitigation by expressing many factors that enhance microglia phagocytic activity [39], which do not represent the primary cause of the disease, per se, but do affect AD time-course and progression rate. The functions of the genes expressed by DAM are first TREM2-independent and later TREM2-dependent [39], and the transition to fully-activated DAM does not occur in the absence of TREM2 receptors. Increased expression of TREM2 during this stage is a defensive factor linked to Aβ clearance [39,189], indicating that TREM2 is necessary to support phagocytosis at a late stage of the disease. In more advanced stages of the disease, TREM2-expressing microglia, interacting with accumulating neurofibrillary tangles, cause extensive inflammation and neurodegeneration [190,191], but the absence of TREM2 in microglia at this stage of AD, but not at earlier stages, exacerbates AD symptomatology [192,193]. Complement C3, responsible for excessive release of pro-inflammatory mediators and induction of reactive astrocytes [37], is one of the most highly upregulated genes involved in this microglia reaction [194]. Distinct microglia subpopulations, located in different brain areas, seem to have different roles at different times in disease progression [39].
Dark microglia, immunonegative for the homeostatic microglial marker P2RY12 and weakly positive for CX3CR1 and IBA1, were identified from their condensed cytoplasm and nucleoplasm, and for their dilated endoplasmic reticulum and altered mitochondria, which make them distinguishable from typical microglia. Dark microglia interact with blood vessels, axon terminals, and dystrophic dendritic spines, and are highly immunoreactive for CD11b and TREM2 [195]. Dark microglia are found near fibrillar Aβ, near Aβ plaques and dystrophic neurites in the CA1 stratum lacunosum moleculare of the ventral hippocampus of the transgenic strain APP/PS1 [196,197]. Nevertheless, the exact role of dark microglia in the pathogenesis of AD remains unclear [195].
In the hippocampus, microglia have a high “immune-vigilant” phenotype that can be responsible for their higher activation in response to Aβ plaque formation, giving rise to a harmful chronic inflammatory response [183]. Furthermore, hippocampal microglia display lower expression of many proteins, including CXCR3 [185], one receptor involved in neuron–microglia communication, and in microglia recruitment, neuronal reorganization [186], and microglia activation during demyelination [187], as described above. Therefore, decreased levels of the CXCR3 receptor and other proteins in AD-vulnerable brain regions may impair microglia response and recruitment.
Microglia express α7nAChR [198] that possibly drive the “cholinergic anti-inflammatory pathway” that regulates systemic inflammatory responses [199]. In CA1 and CA3 of TGCRND8 mice, a transgenic model of AD, microglia surrounding Aβ plaques (plaque activated microglia, PAM) have a different phenotype and show differential expression of α7nAChR (Figure 3). In TGCRND8 mice, microglia in CA1 are bigger, more reactive, and express higher levels of α7nAChR than in WT mice and in CA3 of Tg mice (Figure 3). In CA3, microglia show a ramified state and express α7nAChR (Figure 3B), while in CA1 microglia have a round cell body with shorter branching, and the expression of α7nAChR is more intense (Figure 3C). This differential response of microglia in CA1 and CA3 around plaques confirms one more time the differential spatial states of the cells. Hart and colleagues [200] showed a further regional difference between microglia located in the white matter versus microglia located in the grey matter [184]. Region-specific variations in gene expression (both increases and decreases) may be implicated in the progression or in the resolution of neurodegenerative diseases [201].
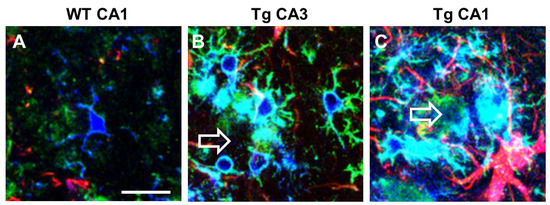
Figure 3. Intense upregulation of α7AChNR in reactive plaque-associated microglia in CA1 and CA3 of TgCRND8 mice. Comparison with WT mice. Anti IBA1 antibody (blue), anti α7AChNR antibody (green), anti GFAP antibody (red). Empty arrows indicate Aβ plaques. (A) A microglia cell with faint α7AChNR immunofluorescence in CA1 hippocampus of WT mouse. (B) Ramified plaque-associated microglia with intense α7AChNR immunofluorescence in CA3 of a TgCRND8 mouse. (C) Amoeboid plaque-associated microglia with very intense α7AChNR immunofluorescence in CA1 of a TgCRND8 mouse. Astrocytes are also visible in red. Scale bar: 20 µm (from [202]).
In the hippocampus of AD patients, the subregional pattern of atrophy is different from other groups of neurodegenerative conditions [203,204], and understanding the molecular and cellular mechanisms that lead to such a subregional vulnerability could unveil therapeutic strategies to alleviate the progression of memory decline. Genome-wide association studies (GWAS) identified AD onset risk loci that are associated with genes involved in microglia physiology and responses, such as CR1 (complement receptor type 1), SPI1 (transcription factor PU.1), TREM2 (triggering receptor expressed on myeloid cells 2), and CD33 [205].
Boosting microglia defensive capabilities with cell-specific therapies may offer new avenues for preventing or reversing neurodegeneration. Further work is needed to demonstrate and dissect these features.
This entry is adapted from the peer-reviewed paper 10.3390/ijms241813668
This entry is offline, you can click here to edit this entry!