Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Neurosciences
Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal dementia (FDT) are progressive neurodegenerative disorders that, in several cases, overlap in clinical presentation, and genetic and pathological disease mechanisms. About 10–15% of ALS cases and up to 40% of FTD are familial, usually with dominant traits. ALS and FTD, in several cases, share common gene mutations, such as in C9ORF72, TARDBP, SQSTM-1, FUS, VCP, CHCHD10, and TBK-1.
- genetic
- amyotrophic lateral sclerosis
- frontotemporal dementia
- immune system
- neuroinflammation
1. Genetic in the ALS-FTD Continuum
During the last few years, several mutations in genes related to ALS and FTD were discovered due to improvements in genetic sequencing analysis and their ease of use [24].
Superoxide dismutase 1 (SOD1) is the primary gene-related only to ALS development [25]. It was discovered in 1993, with more than 200 different mutations described. Although SOD1 mutations are highly heterogeneous, symptoms are often similar to sporadic forms of ALS. In some cases, patients with SOD1 mutations present a more extended disease duration and a pure motor phenotype, generally starting from the lower limbs [26]. Other genes implicated almost exclusively in ALS are the NIMA Related Kinase 1 (NEK1), vesicle-associated membrane protein-associated protein B (VAPB), angiogenin (ANG), senataxin (SETX), and alsin (ALS2) [27,28,29].
Granulin (GRN) and microtubule-associated protein tau (MAPT) are the primary genes related to FTD [30,31]. GRN gene mutations were first identified in families with FTD (5–20%), mainly as autosomal dominant forms associated with chromosome 17. Until now, more than 120 GRN mutations have been identified, excluding most missense variants of GRN that are risk factors for Alzheimer’s disease (AD) rather than FTD [32]. Heterozygous mutations in GRN cause FTD, while homozygous mutations cause a lysosomal storage disorder named neuronal ceroid lipofuscinosis, suggesting that progranulin is involved in lysosomal biogenesis and homeostasis [33].
The MAPT gene, identified more than 40 years ago, is located on chromosome 17q21 and encodes for tau protein, a protein expressed in neurons of several brain regions and less in glial cells [34]. Until now, more than 100 MAPT mutations have been identified [35]. From a pathological point of view, mutations in MAPT occur primarily in the microtubule-binding repeat domain, and consequently, tau has less affinity to microtubules [35,36]. Other minor gene mutations exclusively associated with FTD and not ALS were found on CHMP2B, CCNF, and TIA1 [15,16].
Some gene mutations are detectable in the ALS-FTD spectrum disorder and include C9ORF72, TAR DNA-binding protein (TARDBP), Sequestosome-1 (SQSTM-1), Fused in sarcoma (FUS), Valosin containing protein (VCP), Coiled-coil-helix-coiled-coil-helix domain containing 10 (CHCHD10), Optineurin (OPTN) and Tank-binding kinase 1 (TBK-1) [37,38,39,40,41,42]. The first genetic link between ALS and FTD was the discovery of the TARDBP mutation [43]. Mutations involving TARDBP act on TDP-43 protein, a DNA/RNA binding protein physiologically involved in RNA metabolism. For TARDBP, more than 70 different genetic variants have been described [39,41]. Mutations in this gene can act with two possible pathogenic mechanisms: the first is a depletion of this protein, leading to a loss of function, while the second is a gain of function of the aggregates in terms of toxicity [36].
The hexanucleotide repeat expansion within C9ORF72 on chromosome 9p21 was identified in 2011, and it is responsible for a high percentage of familial cases of ALS and FTD, especially when behavioral symptoms are present [44,45]. Discovered for the first time associated with ALS-FTD cases, it has also been associated with AD, Parkinson’s Disease, atypical parkinsonism, and Huntington’s disease. Although the exact function of C9Orf72 is yet to be defined, the pathological degeneration related to this gene is provoked by several mechanisms, such as accumulation of RNA containing GGGGCC repeat in the brain and spinal cord, direct haploinsufficiency of C9Orf72 protein, dipeptide repeat protein toxicity arising from repeat-associated non-AUG translation occurring off the expansion, and disruption of nucleocytoplasmic transport [38,46,47,48,49].
SQSTM1 gene is located on 5q35 and encodes p62, a multifunctional protein involved in various cellular functions related to neurodegenerative processes, including apoptosis, the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling, ubiquitin-mediated autophagy, and transcription regulation. Defective p26 accumulates in aggregates, disturbing selective autophagy pathways [50].
Among the most recently discovered genes, in 2015, TBK1 was described as associated with ALS-FTD in roughly 1% of European cases. TBK1 is a member of the IkB kinase family involved in autophagy, mitophagy, and innate immune signaling. Most mutations are loss of function due to deletion of the C-terminal domain leading to activation of autophagy and inflammatory pathways via interferon (IFN) type 1. Even with high variability in age at onset and disease duration, clinically, TBK1 mutations are associated mainly with bulbar onset ALS and cognitive impairment [40,51,52,53].
VCP represented another rare early example of gene overlapping; to date, more than 30 mutations have been reported [54]. Many of them are located on exon five within the N-terminal CDC48 domain, which is involved in ubiquitin-binding, meaning that mutations in this region may negatively affect the ubiquitin protein degradation pathway [55]. From a clinical point of view, mutations in VCP are associated with several disorders, Paget’s disease and inclusion body myositis among them [54]. Patients with ALS-FTD carriers of VCP mutations are phenotypically similar to sporadic forms [54].
Mutations in OPTN, encoding optineurin, are rare and associated with both neurological and non-neurological conditions, including the ALS-FTD continuum [56]. OPTN was identified for the first time as an ALS causative gene in 2010, and since then, more than 20 mutations have been reported. The involvement of OPTN in FTD is less clear, although the case series reported a percentage of 5% of FTD patients carrying the OPTN mutation [57]. Optineurin is a protein involved in several cellular processes that act on several pathways. Importantly, OPTN influences the innate immune response by negatively regulating inflammatory pathways [58].
Lastly, mutations in the CHCHD10 gene encode for a small percentage of ALS-FTD patients. CHCHD10 is a mitochondrial protein that regulates mitochondrial metabolism, synthesizes respiratory chain components, and modulates cell apoptosis [59]. At least 30 variants have since been reported, and they are concentrated on exon two of the gene encoding the non-structured N-terminal [59]. In addition to ALS-FTD, disorders related to CHCHD10 mutations are several and varied, such as myopathy, Charcot-Marie Tooth neuropathy, and cerebellar ataxia [60,61].
The involved genes along the ALS-FTD continuum are summarized in Figure 1.
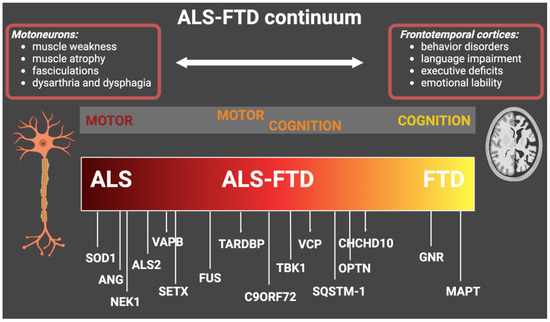
Figure 1. Genes with genetic mutations that cause ALS, ALS-FTD, and FTD. The figure shows the main causative genes associated with the ALS-FTD continuum and the main related clinical features. ALS: amyotrophic lateral sclerosis; FTD: frontotemporal dementia.
2. Autoimmune Disorders and Systemic Inflammation in Genetic ALS/FTD
Several clinical studies focused on the epidemiology of sporadic ALS and FTD support the idea that patients affected by these disorders have a higher incidence of autoimmune diseases. In 2013, the group of Turner evaluated a large UK cohort with autoimmune diseases (e.g., asthma, celiac disease, young-onset diabetes, multiple sclerosis, and systemic lupus erythematosus), reporting in this group a higher percentage of ALS compared to the general population and hypothesizing the possibility of shared genetic and/or environmental risk factors [161]. Although the data is less evident in cohorts of sporadic FTD patients [162], an interesting paper investigated the association between immune disorders and patients with mutations in the GRN gene, observing a significantly increased risk of autoimmune disorders clustered around inflammatory arthritis, cutaneous and gastrointestinal disorders in the PGNR cohort [163].
Regarding other selected cohorts of genetic patients, more results regard the C9Orf72 patients. Firstly, in 2016, in a US cohort, it was reported a high prevalence of autoimmune diseases (similar to what was described for GRN patients) in C9Orf72 ALS/FTD patients compared to controls and other sporadic neurodegenerative patients [164]. A recent multicentric Italian cohort study described 150 ALS C9Orf72 patients, in which an increased incidence of autoimmune diseases was not observed, whereas the authors reported a longer survival in C9Orf72 patients with ALS and thyroid disorders [17]. Looking at the problem in reverse, genotyping a small cohort of patients with ALS and multiple sclerosis, 80% of patients with both neurological conditions carried a C9Orf72 mutation. In the same report, they described an imbalance in the immune pathway, with an NF-κB activation and a concomitant CXCL10 downregulation in the CSF [165]. The presented studies and the possible correlation between NDDs, genetics, and autoimmunity are supported by the recent detection of TBK1 mutations in the ALS/FTD spectrum. In fact, as we have mentioned, TBK1 plays a crucial role as the hub for many innate immune signaling pathways [166].
Also, while systemic inflammation is overall present in ALS/FTD patients, it is not clear if these phenomena are a disease consequence or have a causative role. As global markers of inflammation and peripheral immune disbalance, several recent studies focused on alterations in peripheral markers, such as the neutrophil-to-lymphocyte ratio, in ALS and FTD. Alterations in this ratio, and others related, were associated with poorer prognosis and shorter survival by several groups [167,168,169], but in no study, the genetic component is considered. Also, the studies that analyzed the C-reactive protein in ALS did not primarily focus on genetic subgroups of patients [170,171].
This entry is adapted from the peer-reviewed paper 10.3390/genes14081658
This entry is offline, you can click here to edit this entry!