All the cells of an organism contain the same genome. However, each cell expresses only a minor fraction of its potential and, in particular, the genes encoding the proteins necessary for basal metabolism and the proteins responsible for its specific phenotype. The ability to use only the right and necessary genes involved in specific functions depends on the structural organization of the nuclear chromatin, which in turn depends on the epigenetic history of each cell, which is stored in the form of a collection of DNA and protein modifications. Among these modifications, DNA methylation and many kinds of post-translational modifications of histones play a key role in organizing the complex indexing of usable genes.
1. Introduction
Since Gurdon’s experiments [
1], based on the transplantation into enucleated oocytes of nuclei purified from somatic cells, it became clear that during differentiation, cells do not lose DNA and thus maintain a nucleus with intact potential to generate an entire organism. Despite the presence of the entire genome, however, each specialized cell expresses only a very small percentage of its genome. The reason for specific gene selection lies in the tridimensional organization of chromatin, which is a complex of DNA and proteins. Among these latter molecules, the most represented are histones, i.e., basic proteins that are highly conserved in evolution, which interact with DNA, allowing condensation of the nuclear genome in the very small volume of eukaryotic nuclei. The first level of chromatin organization is the nucleosome, in which about 147 base pairs (bp) of DNA are wrapped around a protein octamer formed by two molecules of each of the histones H2A, H2B, H3, and H4 (called “core” histones”). A fifth histone, the “linker” H1 histone then seals together the points at which DNA enters and exits the nucleosome. Interaction among H1 molecules allows the formation of more condensed DNA fibers [
2,
3,
4,
5,
6,
7,
8].
Most importantly, the chromatin structure differs at the level of different genes that, depending on their structural organization, can be transcriptionally repressed or active, and this is why different cells are able to express different and specific genes, as well as a family of common genes involved in basic metabolism. Thus, specific properties and the behavior of somatic cells do not depend on changes in the genotype but on the specific arrangement of chromatin, according to the concept of “epigenetics” proposed by Waddington many decades ago [
9], which is now widely accepted [
4,
10].
The chromatin structure is highly dynamic and changes in different cells during development and differentiation; it also changes in terminally differentiated cells in response to specific inducing factors, such as, for example, thyroid or steroid hormones [
11]. At least three connected mechanisms are known to induce structural rearrangements of chromatin (
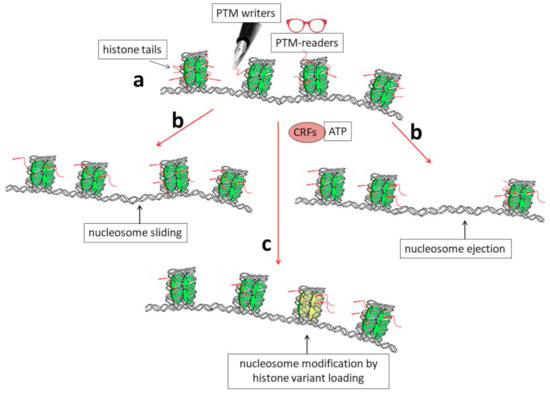
Figure 1): (i) post-translational modification (PTM) of histone proteins [
10,
12] together with DNA methylation [
10,
13,
14]; (ii) the activity of ATP-dependent complexes that are able to induce modifications in the structure/position of nucleosomes [
15], and, finally, (iii) the synthesis and incorporation of histone variants into chromatin [
7,
16,
17,
18].
Figure 1. Schematic representation showing the biochemical mechanisms that allow dynamic modifications of chromatin organization. (a) Histone tails protrude from nucleosomes (red segments) and can be the targets of enzymes (PTM writers) that are able to introduce into them post-translational modifications (PTMs). These modifications can, in turn, directly affect DNA–histone interactions and/or allow chromatin structural modifications through the binding of other specific factors (PTM readers). (b) A modification of the nucleosome position by sliding or nucleosome ejection can be catalyzed by ATP-dependent chromatin-remodeling factors (CRFs). (c) Finally, the incorporation of histone variants into chromatin, such as H3.3, can induce structural and functional modifications of nucleosomes (shown as the yellow nucleosome core particle in the figure).
2. General Properties of Genes Encoding Histone Variants
Although highly conserved in evolution, and thus among different species, histone proteins exist under different isoforms in the same species. These isoforms seem to have different effects on chromatin structure and then on gene expression.
Interestingly, genes encoding the main histone species differ both in structure and expression from those encoding variants synthesized only in specific moments of cell differentiation. In particular, the main histone species (also known as replication-dependent or canonical histones) are synthesized exclusively during the S phase of the cell cycle when DNA is replicated. The corresponding genes are highly repeated and very often clustered; moreover, they do not contain introns, and the corresponding mRNAs are not poly-adenylated [
19,
20,
21]. All these features are related to the necessity of having mRNAs that are immediately available for translation and then for degradation. On the other hand, genes encoding constitutive/variant histones (also known as replication-independent or non-canonical histones) are similar to all the other genes in that they are mostly unique genes that are transcribed, independently of DNA replication, into mRNAs that can contain introns and are polyadenylated [
22]. These genes should be regulated during differentiation in order to produce proteins that are able to bind specific regions of chromatin, thus allowing activation/repression of specific genes.
Many years ago, the group showed that two histone variants, i.e., the linker histone H1.0 and the core histone H3.3, are specifically expressed in the rat brain during brain maturation [
23]. Interestingly, a combination of run-on experiments on isolated nuclei and transcription inhibition using actinomycin D demonstrated that the two genes have an “open” structure and that H1.0 and H3.3 histone synthesis in the central nervous system (CNS) is largely regulated at the post-transcriptional level [
24]. Indeed, people identified a group of proteins that are able to bind their mRNAs and cloned a couple of them (CSD-C2/PIPPin and LPI/PEP-19) [
25,
26,
27,
28,
29]. Notably, in a very recent and interesting paper focused on the effects of hunger on neuronal histone modifications and the life span of the
Drosophila fruit fly, the effects of a diet containing low amounts of branched-chain amino acids (BCAAs) were analyzed. In particular, the authors found that total histone H3 abundance decreased in flies fed a low BCAA diet, while H3 mRNA increased [
30]. This observation suggested that post-transcriptional events might be of importance for general H3 metabolism. Moreover, the paper also reported that canonical H3 is evicted from chromatin and replaced with H3.3 [
30].
It is also worth noting that two genes encoding the H3.3 histone are present in mammals:
H3.3A and
H3.3B (also called
H3F3A and
H3F3B), which are located on different chromosomes. While the distribution of exons and introns, as well as promoters and other regulatory regions, are different in the two genes, the corresponding proteins are identical [
31] and also highly conserved in evolution. Thus, it is highly probable that the existence of the two genes is not important for having two proteins with different activities but, instead, because it offers the possibility to regulate the genes (and the related mRNAs) independently and/or with different mechanisms [
32]. In particular, it was suggested that the two genes may have cell type-specific expression [
33], although their overall activity in different tissues was reported to be quite similar [
34].
Actually, the H3.3 protein is not so different with respect to the canonical H3.1 and H3.2 isoforms: indeed, it differs by only five and four amino acids, respectively, from them [
35,
36,
37]. However, these amino acids are, for example, sufficient to allow H3.3 to interact with specific histone chaperones, such as the Death domain-associated protein (DAXX), the alpha-thalassemia/mental retardation X-linked protein (ATRX) complex, and the histone regulator A (Hira)/calcineurin-binding protein 1 (Cabin 1)/ubinuclein1 (Ubn1) complex, involved in its loading on chromatin [
7,
38,
39,
40,
41]. The H3.3 interaction with these chaperones is determinant for its deposition on specific regions of the genome. It was shown, for example, that a mutation of the ATRX complex leads, as a consequence, to variation in the deposition of H3.3 and chromatin accessibility in association with an alteration in gene expression [
42].
The H3.3 variant is indeed very often bound to active chromatin and regulates transcription. In 2002, Ahmad and Henikoff reported that in
Drosophila cells, H3.3 is the only H3 species deposited in chromatin in a replication-independent way, and they suggested that this event might be responsible for the activation of genes previously silenced because of histone PTMs [
43]. As a confirmation of its gene-activating function, the H3.3 histone was also found at the level of active enhancers [
44,
45,
46].
It was also reported that some chromatin remodeling complexes, such as the SWItch/Sucrose Non-Fermentable (SWI/SNF) complex and, in particular, its subunit “T-rich interactive domain-containing protein 1A” (ARAD1A) are required for maintaining the H3.3 histone at the level of regulatory sequences, among which are the so-called super-enhancers [
47]. On the other hand, by interacting with the ATRX/DAXX chaperones, H3.3 might also be loaded on pericentric heterochromatin and telomeres [
38,
48,
49]. This event, together with lysine 56 (H3.3K56) acetylation, seems necessary for chromosome segregation in mammals. Indeed, it was shown that cell lines carrying the mutation K56R increase cell death and modify cell morphology [
50].
Recently, a brain-specific function of the chromodomain-helicase-DNA binding protein 1 (CHD1) which is a member of the SWI/SNF family of chromatin remodeling complexes was reported in
Drosophila. CHD1 is indeed involved in the loading of H3.3 in the fly brain, where it seems to contribute to the regulation of genes that control the homeostasis of hunger and satiety signals [
51]. On the other hand, as a demonstration of the wide range of tissues and functions in which H3.3 histone is probably involved, it was also found to be essential for the chromatin transitions that accompany
Drosophila male germline maturation [
52].
Further work is required to understand how H3.3, which, as mentioned above, is not so different from the canonical H3 species, can stimulate transcription. Evidently, its sequence should contain features that are able to attract, directly or indirectly, the transcriptional apparatus to the genes to which it is bound. Interestingly, for example, the H3.3 amino-terminal tail contains a serine residue (S31) that is not present in the other H3 species (which contain, instead, an alanine at that position). Moreover, this serine can be phosphorylated, and it has been suggested that this might represent a feature determinant for preferential transcription [
53]. It should be noted that S31 is actually S32 in the original amino acid sequence of the H3.3 protein; however, in the mature protein, it becomes S31 because the initiator methionine is immediately cleaved during translation ([
54] and references therein). Notably, it was recently demonstrated that S31 phosphorylation can also modify the accessibility of regulatory factors at telomeres during replication, thus stabilizing heterochromatin probably by influencing the activity of histone lysine demethylase 4B (KDM4B) [
55].
One possible link between H3.3 modification and gene expression was suggested by Martire and co-workers [
56]. Using mouse embryonal stem cells (mESCs), these authors showed that the cells missing histone H3.3 cannot normally acetylate the enhancers that are activated during differentiation and, more specifically, show a reduction in the acetylation of the H3 histone at lysine 27. The normal ability to regulate acetylation would depend on the stimulation of p300 acetyltransferase by phosphorylation at specific sites of the H3.3 histone variant [
56,
57].
This entry is adapted from the peer-reviewed paper 10.3390/ijms241311028