Sjögren’s Syndrome (SS) is a chronic autoimmune disorder characterized by focal mononuclear cell infiltrates that surround the ducts of the exocrine glands, impairing the function of their secretory units. Compared to other autoimmune disorders, SS is associated with a notably high incidence of non-Hodgkin lymphoma (NHL) and more frequently mucosa associated lymphoid tissue (MALT) lymphoma, leading to increased morbidity and mortality rates. High risk features of lymphoma development include systemic extraepithelial manifestations, low serum levels of complement component C4 and mixed type II cryoglobulinemia. The discrimination between reactive and neoplastic lymphoepithelial lesion (LEL) is challenging, probably reflecting a continuum in the evolution from purely inflammatory lymphoid infiltration to the clonal neoplastic evolution. Early lesions display a predominance of activated T cells, while B cells prevail in severe histologic lesions. This strong B cell infiltration is not only a morphologic phenomenon, but it is also progressively associated with the presence of ectopic germinal centers (GCs).
- Sjögren’s Syndrome Lymphomagenesis
1. Introduction
Sjögren’s Syndrome (SS) is benign autoimmune disease characterized by organ specific as well as systemic manifestations. Of interest this benign autoimmune disease can potentially lead to lymphomagenesis. A major complication in the progression of SS is lymphoma development, leading to increased mortality of SS patients that develop lymphoma in the course of their disease[1][2].
B cells displaying a marginal zone (MZ) phenotype are detected in the majority of lymphomas developing in the setting of SS. Mucosa associated lymphoid tissue (MALT) lymphomas account for 65% of SS related lymphomas, followed by diffuse large B cell lymphomas (DLBCL) and nodal MZ lymphomas[20][21]. Of note, most cases of DLBCL are probably the evolution of low-grade MZ lymphomas[22][23]. Since MALT lymphomas account for the majority of SS associated lymphomas, the pathophysiologic mechanisms described in this review mainly refer to this type of lymphoma.
Compared to patients with other autoimmune diseases, namely systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA), patients with primary SS (pSS) present a higher risk for lymphoma development[24]. Several clinical and laboratory parameters have been proposed to distinguish patients at this high risk [20][3][4][5][6][7][8][9][10][11][12][13][14][15][16][17][18][19].
SS is characterized by lymphocytic infiltration of the exocrine glands. Lymphoepithelial sialadenitis (LESA), a notable histological feature of SS, is characterized by the presence of lymphoid populations surrounding and infiltrating the salivary ducts, along with disorganization and proliferation of the ductal epithelial cells[25]. The spectrum of histopathologic features of LESA ranges from a fully benign lymphoid infiltrate, sometimes associated with lymphoid follicular structures, that does not display immunoglobulin (Ig) light chain restriction in B-cells, to lymphoproliferative lesions with presence of centrocyte-like cells with or without areas of Ig light chain restriction in B cells[25]. B-cell clones are found in approximately 50% of LESAs without morphological or clinical evidence of lymphoma[26][27]. Monoclonal B cell expansion does not necessarily constitute lymphoma. The evolution from persistent polyclonal B cell activation to monoclonal B cell expansion and thereafter lymphoma development is a multistep process.
2. Summarized Model of Lymphomagenesis in SS
The local auto- antigen expression by the epithelium of the LEL in SGs of SS patients drives the emergence of autoreactive B-cell clones bearing an RF reacting BCR. Opsonized epithelial apoptotic particles and ICs containing nuclear autoantigens bind to plasmacytoid DCs (pDCs) and RF B cells via TLRs and Fc receptors, respectively. TLR activation in pDCs leads to IFNa release that subsequently stimulates myeloid DCs (mDCs) to produce BAFF. The same apoptotic particles and ICs also bind to RF B cells via TLR and BCR, respectively. TLR activation in B cells results in enhanced BCR mediated signaling and upregulation of BAFF-R. BAFF, secreted by mDCs, further upregulates TLR expression, favors B cell survival, promotes immunoglobulin class-switching and plasma cell differentiation (Figure 1, Z B cell maturation pathway). The activated mDCs also act as antigen presenting cells to T cells that help B cell responses (Figure 1, GC B cell maturation pathway). Therefore, the combined signals of BCR, TLR, BAFF and IFNa drive the propagated production of autoantibodies and lead to expansion of RF B cells (Figure 1)[28].
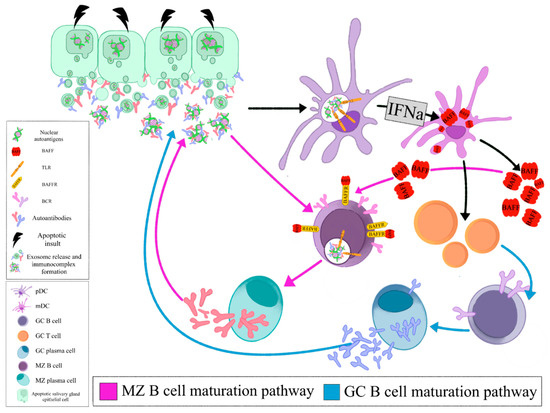
Figure 1. Opsonized epithelial apoptotic particles and ICs containing nuclear autoantigens bind to plasmacytoid DCs (pDCs) and RF B cells via TLRs and Fc receptors, respectively. TLR activation in pDCs leads to IFNa release that subsequently stimulates myeloid DCs (mDCs) to produce BAFF. The same apoptotic particles and ICs also bind to RF B cells via TLR and BCR, respectively. TLR activation in B cells results in enhanced BCR mediated signaling and upregulation of BAFF-R. BAFF, secreted by mDCs, further upregulates TLR expression, favors B cell survival, promotes immunoglobulin class- switching and plasma cell differentiation (MZ B cell maturation pathway). The activated mDCs also act as antigen presenting cells to T cells that help B cell responses (GC B cell maturation pathway). (pDC: plasmacytoid dendritic cell, mDC: myeloid dendritic cell, GC: germinal center, MZ: marginal zone, BAFF: B cell activating factor, TLR: Toll like receptor, BAFFR: BAFF receptor, BCR: B cell receptor, IFNa: interferon a).
Selection of these RF-expressing B cells offers them a proliferative advantage, leading to their malignant transformation and eventually MALT lymphoma development. Autoreactive MZ B cells have been shown to be negatively selected in healthy individuals. RF-expressing, autoreactive MZ like B cells in SS bypass the peripheral checkpoint against autoreactivity and proceed to proliferation and differentiation through T cell independent pathways[29].
Lymphoma development in the setting of SS is associated with increased overall disease mortality. A multistep process leads to the transition of reactive LESA to lymphomagenesis. Chronic antigenic stimulation leads to abnormal B cell activation in the SG glands of SS patients and the emergence of autoreactive B cell clones. Loss of immune control, ectopic GC formation and oncogenic events further drive the malignant transformation to lymphoma (Figure 2).
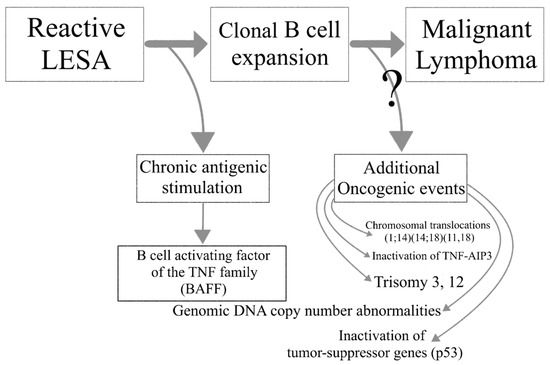
Figure 2. The transition from lymphoepithelial sialadenitis to malignant lymphoma.
This entry is adapted from the peer-reviewed paper 10.3390/jcm9123794
References
- Voulgarelis, M.; Ziakas, P.D.; Papageorgiou, A.; Baimpa, E.; Tzioufas, A.G.; Moutsopoulos, H.M. Prognosis and outcome of non-Hodgkin lymphoma in primary Sjögren syndrome. Medicine 2012, 91, 1-9, doi:10.1097/md.0b013e31824125e4.
- Sikara, M.; Ziogas, D.; Argyropoulou, O.; Papageorgiou, A.; Tzioufas, A.; Voulgarelis, M. SAT0208 TEN-YEAR OVERALL SURVIVAL AND STANDARIZED MORTALITY RATIO IN THE LARGEST SINGLE CENTER COHORT OF PATIENTS WITH PRIMARY SJOGREN’S ASSOCIATED LYMPHOMAS. Annals of the rheumatic diseases 2019, 78, 1179-1179, doi:10.1136/annrheumdis-2019-eular.5794.
- Skopouli, F.N.; Dafni, U.; Ioannidis, J.P.; Moutsopoulos, H.M. Clinical evolution, and morbidity and mortality of primary Sjögren's syndrome. Seminars in arthritis and rheumatism 2000, 29, 296-304, doi:10.1016/s0049-0172(00)80016-5.
- Ioannidis, J.P.; Vassiliou, V.A.; Moutsopoulos, H.M. Long-term risk of mortality and lymphoproliferative disease and predictive classification of primary Sjögren's syndrome. Arthritis and rheumatism 2002, 46, 741-747, doi:10.1002/art.10221.
- Papageorgiou, A.; Voulgarelis, M.; Tzioufas, A.G. Clinical picture, outcome and predictive factors of lymphoma in Sjӧgren syndrome. Autoimmun Rev 2015, 14, 641-649, doi:10.1016/j.autrev.2015.03.004.
- Fragkioudaki, S.; Mavragani, C.P.; Moutsopoulos, H.M. Predicting the risk for lymphoma development in Sjogren syndrome: An easy tool for clinical use. Medicine 2016, 95, e3766, doi:10.1097/md.0000000000003766.
- Sutcliffe, N.; Inanc, M.; Speight, P.; Isenberg, D. Predictors of lymphoma development in primary Sjögren's syndrome. Seminars in arthritis and rheumatism 1998, 28, 80-87, doi:https://doi.org/10.1016/S0049-0172(98)80040-1.
- Baimpa, E.; Dahabreh, I.J.; Voulgarelis, M.; Moutsopoulos, H.M. Hematologic manifestations and predictors of lymphoma development in primary Sjögren syndrome: clinical and pathophysiologic aspects. Medicine 2009, 88, 284-293, doi:10.1097/MD.0b013e3181b76ab5.
- Theander, E.; Henriksson, G.; Ljungberg, O.; Mandl, T.; Manthorpe, R.; Jacobsson, L.T. Lymphoma and other malignancies in primary Sjögren's syndrome: a cohort study on cancer incidence and lymphoma predictors. Annals of the rheumatic diseases 2006, 65, 796-803, doi:10.1136/ard.2005.041186.
- Risselada, A.P.; Kruize, A.A.; Bijlsma, J.W. Clinical features distinguishing lymphoma development in primary Sjögren's Syndrome--a retrospective cohort study. Seminars in arthritis and rheumatism 2013, 43, 171-177, doi:10.1016/j.semarthrit.2013.03.001.
- Nocturne, G.; Virone, A.; Ng, W.-F.; Le Guern, V.; Hachulla, E.; Cornec, D.; Daien, C.; Vittecoq, O.; Bienvenu, B.; Marcelli, C., et al. Rheumatoid Factor and Disease Activity Are Independent Predictors of Lymphoma in Primary Sjögren's Syndrome. Arthritis & Rheumatology 2016, 68, 977-985, doi:10.1002/art.39518.
- Quartuccio, L.; Isola, M.; Baldini, C.; Priori, R.; Bartoloni Bocci, E.; Carubbi, F.; Maset, M.; Gregoraci, G.; Della Mea, V.; Salvin, S., et al. Biomarkers of lymphoma in Sjögren's syndrome and evaluation of the lymphoma risk in prelymphomatous conditions: results of a multicenter study. Journal of autoimmunity 2014, 51, 75-80, doi:10.1016/j.jaut.2013.10.002.
- Retamozo, S.; Gheitasi, H.; Quartuccio, L.; Kostov, B.; Corazza, L.; Bové, A.; Sisó-Almirall, A.; Gandía, M.; Ramos-Casals, M.; De Vita, S., et al. Cryoglobulinaemic vasculitis at diagnosis predicts mortality in primary Sjögren syndrome: analysis of 515 patients. Rheumatology 2016, 55, 1443-1451, doi:10.1093/rheumatology/kew194.
- Solans-Laqué, R.; López-Hernandez, A.; Bosch-Gil, J.A.; Palacios, A.; Campillo, M.; Vilardell-Tarres, M. Risk, predictors, and clinical characteristics of lymphoma development in primary Sjögren's syndrome. Seminars in arthritis and rheumatism 2011, 41, 415-423, doi:10.1016/j.semarthrit.2011.04.006.
- Tomi, A.-L.; Belkhir, R.; Nocturne, G.; Desmoulins, F.; Berge, E.; Pavy, S.; Miceli, C.; Mariette, X.; Seror, R. Monoclonal gammopathy and risk of lymphoma and multiple myeloma in patients with primary Sjögren's syndrome. Arthritis & Rheumatology 2015, 68, n/a-n/a, doi:10.1002/art.39534.
- Nishishinya, M.B.; Pereda, C.A.; Muñoz-Fernández, S.; Pego-Reigosa, J.M.; Rúa-Figueroa, I.; Andreu, J.L.; Fernández-Castro, M.; Rosas, J.; Loza Santamaría, E. Identification of lymphoma predictors in patients with primary Sjögren's syndrome: a systematic literature review and meta-analysis. Rheumatology international 2015, 35, 17-26, doi:10.1007/s00296-014-3051-x.
- Tzioufas, A.G.; Boumba, D.S.; Skopouli, F.N.; Moutsopoulos, H.M. Mixed monoclonal cryoglobulinemia and monoclonal rheumatoid factor cross-reactive idiotypes as predictive factors for the development of lymphoma in primary Sjögren's syndrome. Arthritis and rheumatism 1996, 39, 767-772, doi:10.1002/art.1780390508.
- Theander, E.; Vasaitis, L.; Baecklund, E.; Nordmark, G.; Warfvinge, G.; Liedholm, R.; Brokstad, K.; Jonsson, R.; Jonsson, M.V. Lymphoid organisation in labial salivary gland biopsies is a possible predictor for the development of malignant lymphoma in primary Sjögren's syndrome. Annals of the rheumatic diseases 2011, 70, 1363-1368, doi:10.1136/ard.2010.144782.
- Risselada, A.P.; Kruize, A.A.; Goldschmeding, R.; Lafeber, F.P.; Bijlsma, J.W.; van Roon, J.A. The prognostic value of routinely performed minor salivary gland assessments in primary Sjögren's syndrome. Annals of the rheumatic diseases 2014, 73, 1537-1540, doi:10.1136/annrheumdis-2013-204634.
- Papageorgiou, A.; Ziogas, D.C.; Mavragani, C.P.; Zintzaras, E.; Tzioufas, A.G.; Moutsopoulos, H.M.; Voulgarelis, M. Predicting the outcome of Sjogren's syndrome-associated non-hodgkin's lymphoma patients. PLoS One 2015, 10, e0116189, doi:10.1371/journal.pone.0116189.
- Ekström Smedby, K.; Vajdic, C.M.; Falster, M.; Engels, E.A.; Martínez-Maza, O.; Turner, J.; Hjalgrim, H.; Vineis, P.; Seniori Costantini, A.; Bracci, P.M., et al. Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: a pooled analysis within the InterLymph Consortium. Blood 2008, 111, 4029-4038, doi:10.1182/blood-2007-10-119974.
- Maeshima, A.M.; Taniguchi, H.; Toyoda, K.; Yamauchi, N.; Makita, S.; Fukuhara, S.; Munakata, W.; Maruyama, D.; Kobayashi, Y.; Tobinai, K. Clinicopathological features of histological transformation from extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue to diffuse large B-cell lymphoma: an analysis of 467 patients. British Journal of Haematology 2016, 174, 923-931, doi:10.1111/bjh.14153.
- Gorodetskiy, V.R.; Probatova, N.A. Clonal relationship of marginal zone lymphoma and diffuse large B-cell lymphoma in Sjogren's syndrome patients: case series study and review of the literature. 2020, 40, 499-506, doi:10.1007/s00296-019-04470-x.
- Zintzaras, E.; Voulgarelis, M.; Moutsopoulos, H.M. The risk of lymphoma development in autoimmune diseases: a meta-analysis. Arch Intern Med 2005, 165, 2337-2344, doi:10.1001/archinte.165.20.2337.
- DiGiuseppe, J.A.; Corio, R.L.; Westra, W.H. Lymphoid infiltrates of the salivary glands: pathology, biology and clinical significance. Current opinion in oncology 1996, 8, 232-237, doi:10.1097/00001622-199605000-00011.
- Carbone, A.; Gloghini, A.; Ferlito, A. Pathological Features of Lymphoid Proliferations of the Salivary Glands: Lymphoepithelial Sialadenitis versus Low-Grade B-Cell Lymphoma of the Malt Type. Annals of Otology, Rhinology & Laryngology 2000, 109, 1170-1175, doi:10.1177/000348940010901217.
- Pijpe, J.; Kalk, W.W.I.; van der Wal, J.E.; Vissink, A.; Kluin, P.M.; Roodenburg, J.L.N.; Bootsma, H.; Kallenberg, C.G.M.; Spijkervet, F.K.L. Parotid gland biopsy compared with labial biopsy in the diagnosis of patients with primary Sjögren's syndrome. Rheumatology 2007, 46, 335-341, doi:10.1093/rheumatology/kel266.
- Moisini, I.; Davidson, A. BAFF: a local and systemic target in autoimmune diseases. Clinical & Experimental Immunology 2009, 158, 155-163, doi:10.1111/j.1365-2249.2009.04007.x.
- Hansen, A.; Lipsky, P.E.; Dörner, T. B cells in Sjögren's syndrome: indications for disturbed selection and differentiation in ectopic lymphoid tissue. Arthritis research & therapy 2007, 9, 218-218, doi:10.1186/ar2210.