Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in children and adolescents has generated repercussions, especially a few weeks after infection, for symptomatic patients who tested positive, for asymptomatic ones, or even just the contacts of an infected person, and evolved from severe forms such as multisystem inflammatory syndrome in children (MIS-C) to multifarious clinical manifestations in long COVID (LC). Referred to under the umbrella term LC, the onset of persistent and highly heterogeneous symptoms such as fatigue, post-exertion malaise, cognitive dysfunction, and others have a major impact on the child’s daily quality of life for months.
- long COVID
- infection
- inflammation
- children
1. Introduction
2. The Interplay between Main Molecular and Cellular Pathogenic Mechanisms in Pediatric Long COVID
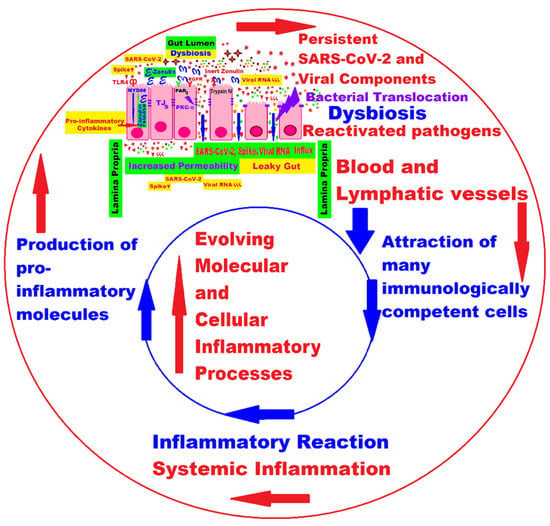
2.1. Persistence of Virus or Viral Components and Prolonged Immune Activation with Inflammation
2.2. Processes Mediated by Autoantibodies in the Spectrum of Autoimmune Diseases
2.3. Long COVID in the Era of the Microbiome and Connected Molecular Mechanisms
This entry is adapted from the peer-reviewed paper 10.3390/ijms241310874
References
- The World Health Organization. International Health Regulations (2005)–Third Edition. Available online: https://www.who.int/publications/i/item/9789241580496 (accessed on 10 April 2023).
- Gostin, L.O.; Katz, R. The International Health Regulations: The Governing Framework for Global Health Security. Milbank Q. 2016, 94, 264–313.
- Wilder-Smith, A.; Osman, S. Public health emergencies of international concern: A historic overview. J. Travel Med. 2020, 27, taaa227.
- Sohrabi, C.; Alsafi, Z.; O’Neill, N.; Khan, M.; Kerwan, A.; Al-Jabir, A.; Iosifidis, C.; Agha, R. World Health Organization declares Global Emergency: A review of the 2019 Novel Coronavirus (COVID-19). Int. J. Surg. 2020, 76, 71–76, Erratum in Int. J. Surg. 2020, 77, 217.
- Zhang, Y.; Yu, B.; Chen, X.; Rich, S.; Mo, Q.; Yan, H. Dynamics of the coronavirus disease 2019 (COVID-19) epidemic in Wuhan City, Hubei Province and China: A second derivative analysis of the cumulative daily diagnosed cases during the first 85 days. Glob. Health J. 2021, 5, 4–11.
- Worobey, M.; Levy, J.I.; Malpica Serrano, L.; Crits-Christoph, A.; Pekar, J.E.; Goldstein, S.A.; Rasmussen, A.L.; Kraemer, M.U.G.; Newman, C.; Koopmans, M.P.G.; et al. The Huanan Seafood Wholesale Market in Wuhan was the early epicenter of the COVID-19 pandemic. Science 2022, 377, 951–959.
- The World Health Organization. Coronavirus disease (COVID-19) Pandemic. Available online: https://www.who.int/europe/emergencies/situations/covid-19 (accessed on 12 April 2023).
- National Library of Medicine; National Center for Biology Information. Severe Acute Respiratory Syndrome Coronavirus 2 Isolate Wuhan-Hu-1, Complete Genome. GenBank: MN908947.3. Available online: https://www.ncbi.nlm.nih.gov/nuccore/MN908947 (accessed on 12 April 2023).
- Grellet, E.; L’Hôte, I.; Goulet, A.; Imbert, I. Replication of the coronavirus genome: A paradox among positive-strand RNA viruses. J. Biol. Chem. 2022, 298, 101923.
- Roland, D.; Gardiner, A.; Razzaq, D.; Rose, K.; Bressan, S.; Honeyford, K.; Buonsenso, D.; Da Dalt, L.; De, T.; Farrugia, R.; et al. Influence of epidemics and pandemics on paediatric ED use: A systematic review. Arch. Dis. Child. 2023, 108, 115–122.
- Pyone, T.; Aung, T.T.; Endericks, T.; Myint, N.W.; Inamdar, L.; Collins, S.; Pwint, K.H.; Hein, B.B.; Wilson, A. Health system governance in strengthening International Health Regulations (IHR) compliance in Myanmar. BMJ Glob. Health 2020, 5, e003566.
- Verikios, G. The dynamic effects of infectious disease outbreaks: The case of pandemic influenza and human coronavirus. Socio-Econ. Plan. Sci. 2020, 71, 100898.
- Castelli, V.; Cimini, A.; Ferri, C. Cytokine Storm in COVID-19: “When You Come Out of the Storm, You Won’t Be the Same Person Who Walked in”. Front. Immunol. 2020, 11, 2132.
- Araf, Y.; Faruqui, N.A.; Anwar, S.; Hosen, M.J. SARS-CoV-2: A new dimension to our understanding of coronaviruses. Int. Microbiol. 2021, 24, 19–24.
- Vahabi, M.; Ghazanfari, T.; Sepehrnia, S. Molecular mimicry, hyperactive immune system, and SARS-COV-2 are three prerequisites of the autoimmune disease triangle following COVID-19 infection. Int. Immunopharmacol. 2022, 112, 109183.
- Keshavarzi, A.; Horry, H.R. Bayesian estimation of a dynamic stochastic general equilibrium model with health disaster risk. Stoch. Environ. Res. Risk Assess. 2023, 37, 1199–1211.
- Cascella, M.; Rajnik, M.; Aleem, A.; Dulebohn, S.C.; Di Napoli, R. Features, Evaluation, and Treatment of Coronavirus (COVID-19). 9 January 2023. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://pubmed.ncbi.nlm.nih.gov/32150360 (accessed on 24 May 2023).
- Zebardast, A.; Hasanzadeh, A.; Ebrahimian Shiadeh, S.A.; Tourani, M.; Yahyapour, Y. COVID-19: A trigger of autoimmune diseases. Cell Biol. Int. 2023, 47, 848–858.
- Vilser, D. Long Covid/Post-COVID-19-Syndrom bei Kindern und Jugendlichen. Pädiatrie 2022, 34, 20–25.
- Castanares-Zapatero, D.; Chalon, P.; Kohn, L.; Dauvrin, M.; Detollenaere, J.; Maertens de Noordhout, C.; Primus-de Jong, C.; Cleemput, I.; Van den Heede, K. Pathophysiology and mechanism of long COVID: A comprehensive review. Ann. Med. 2022, 54, 1473–1487.
- de Carvalho, S.S.; Simões e Silva, A.C.; Sabino, A.D.P.; Evangelista, F.C.; Gomes, K.B.; Dusse, L.M.; Rios, D.R. Influence of ACE I/D Polymorphism on Circulating Levels of Plasminogen Activator Inhibitor 1, D-Dimer, Ultrasensitive C-Reactive Protein and Transforming Growth Factor β1 in Patients Undergoing Hemodialysis. PLoS ONE 2016, 11, e0150613.
- Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol. 2022, 20, 270–284.
- Silva, M.G.; Falcoff, N.L.; Corradi, G.R.; Di Camillo, N.; Seguel, R.F.; Tabaj, G.C.; Guman, G.R.; de Matteo, E.; Nuñez, M.; Gironacci, M.M. Effect of age on human ACE2 and ACE2-expressing alveolar type II cells levels. Pediatr. Res. 2023, 93, 948–952.
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 2010, 84, 12658–12664.
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8.
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105.
- Hachim, I.Y.; Hachim, M.Y.; Talaat, I.M.; López-Ozuna, V.M.; Saheb Sharif-Askari, N.; Al Heialy, S.; Halwani, R.; Hamid, Q. The Molecular Basis of Gender Variations in Mortality Rates Associated with the Novel Coronavirus (COVID-19) Outbreak. Front. Mol. Biosci. 2021, 8, 728409.
- Guo, X.; Zhu, Y.; Hong, Y. Decreased Mortality of COVID-19 With Renin-Angiotensin-Aldosterone System Inhibitors Therapy in Patients with Hypertension: A Meta-Analysis. Hypertension 2020, 76, e13–e14.
- Papola, F.; Biancofiore, V.; Angeletti, C.; Grimaldi, A.; Carucci, A.C.; Cofini, V.; Necozione, S.; Rosciano, A.; Marinangeli, F.; Cervelli, C. Anti-AT1R autoantibodies and prediction of the severity of Covid-19. Hum. Immunol. 2022, 83, 130–133.
- Bastolla, U.; Chambers, P.; Abia, D.; Garcia-Bermejo, M.-L.; Fresno, M. Is Covid-19 Severity Associated with ACE2 Degradation? Front. Drug. Discov. 2022, 1, 789710.
- Matschke, J.; Lütgehetmann, M.; Hagel, C.; Sperhake, J.P.; Schröder, A.S.; Edler, C.; Mushumba, H.; Fitzek, A.; Allweiss, L.; Dandri, M.; et al. Neuropathology of patients with COVID-19 in Germany: A post-mortem case series. Lancet Neurol. 2020, 19, 919–929.
- Asadi-Pooya, A.A.; Akbari, A.; Emami, A.; Lotfi, M.; Rostamihosseinkhani, M.; Nemati, H.; Barzegar, Z.; Kabiri, M.; Zeraatpisheh, Z.; Farjoud-Kouhanjani, M.; et al. Long COVID syndrome-associated brain fog. J. Med. Virol. 2022, 94, 979–984.
- Stephenson, T.; Shafran, R.; Ladhani, S.N. Long COVID in children and adolescents. Curr. Opin. Infect. Dis. 2022, 35, 461–467.
- Griffin, D.E. Why does viral RNA sometimes persist after recovery from acute infections? PLoS Biol. 2022, 20, e3001687.
- Lopez-Leon, S.; Wegman-Ostrosky, T.; Ayuzo del Valle, N.C.; Perelman, C.; Sepulveda, R.; Rebolledo, P.A.; Cuapio, A.; Villapol, S. Long-COVID in children and adolescents: A systematic review and meta-analyses. Sci. Rep. Sci. 2022, 12, 9950.
- Rong, Z.; Mai, H.; Kapoor, S.; Puelles, G.V.; Czogalla, J.; Schädler, J.; Vering, J.; Delbridge, C.; Steinke, H.; Frenzel, H.; et al. SARS-CoV-2 Spike Protein Accumulation in the Skull-Meninges-Brain Axis: Potential Implications for Long-Term Neurological Complications in post-COVID-19. bioRxiv 2023.
- Fullard, J.F.; Lee, H.C.; Voloudakis, G.; Suo, S.; Javidfar, B.; Shao, Z.; Peter, C.; Zhang, W.; Jiang, S.; Corvelo, A.; et al. Single-nucleus transcriptome analysis of human brain immune response in patients with severe COVID-19. Genome Med. 2021, 13, 118.
- Theoharides, T.C. Could SARS-CoV-2 Spike Protein Be Responsible for Long-COVID Syndrome? Mol. Neurobiol. 2022, 59, 1850–1861.
- Theoharides, T.C.; Kempuraj, D. Role of SARS-CoV-2 Spike-Protein-Induced Activation of Microglia and Mast Cells in the Pathogenesis of Neuro-COVID. Cells 2023, 12, 688.
- de Melo, G.D.; Lazarini, F.; Levallois, S.; Hautefort, C.; Michel, V.; Larrous, F.; Verillaud, B.; Aparicio, C.; Wagner, S.; Gheusi, G.; et al. COVID-19-related anosmia is associated with viral persistence and inflammation in human olfactory epithelium and brain infection in hamsters. Sci. Transl. Med. 2021, 13, eabf8396.
- Jacobs, J.J.L. Persistent SARS-2 infections contribute to long COVID-19. Med. Hypotheses 2021, 149, 110538.
- Casabianca, M.; Caula, C.; Titomanlio, L.; Lenglart, L. Neurological consequences of SARS-CoV-2 infections in the pediatric population. Front. Pediatr. 2023, 11, 1123348.
- Tian, Y.; Rong, L.; Nian, W.; He, Y. Review article: Gastrointestinal features in COVID-19 and the possibility of faecal transmission. Aliment. Pharmacol. Ther. 2020, 51, 843–851.
- Ma, X.; Guan, C.; Chen, R.; Wang, Y.; Feng, S.; Wang, R.; Qu, G.; Zhao, S.; Wang, F.; Wang, X.; et al. Pathological and molecular examinations of postmortem testis biopsies reveal SARS-CoV-2 infection in the testis and spermatogenesis damage in COVID-19 patients. Cell. Mol. Immunol. 2021, 18, 487–489.
- Cherne, M.D.; Gentry, A.B.; Nemudraia, A.; Nemudryi, A.; Hedges, J.F.; Walk, H.; Blackwell, K.; Snyder, D.T.; Jerome, M.; Madden, W.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 Is Detected in the Gastrointestinal Tract of Asymptomatic Endoscopy Patients but Is Unlikely to Pose a Significant Risk to Healthcare Personnel. Gastro Hep. Adv. 2022, 1, 844–852.
- Connelly, Z.M.; Whitaker, D.; Dullea, A.; Ramasamy, R. SARS-CoV-2 Effects on the Male Genitourinary System. Am. J. Clin. Exp. Urol. 2022, 10, 199–209.
- Zollner, A.; Koch, R.; Jukic, A.; Pfister, A.; Meyer, M.; Rössler, A.; Kimpel, J.; Adolph, T.E.; Tilg, H. Postacute COVID-19 is Characterized by Gut Viral Antigen Persistence in Inflammatory Bowel Diseases. Gastroenterology 2022, 163, 495–506.e8.
- Arostegui, D.; Castro, K.; Schwarz, S.; Vaidy, K.; Rabinowitz, S.; Wallach, T.D. Persistent SARS-CoV-2 Nucleocapsid Protein Presence in the Intestinal Epithelium of a Pediatric Patient 3 Months After Acute Infection. JPGN Rep. 2022, 3, e152.
- Tullie, L.; Ford, K.; Bisharat, M.; Watson, T.; Thakkar, H.; Mullassery, D.; Giuliani, S.; Blackburn, S.; Cross, K.; De Coppi, P.; et al. Gastrointestinal features in children with COVID-19: An observation of varied presentation in eight children. Lancet Child Adolesc. Health 2020, 4, e19–e20.
- Colmenero, I.; Santonja, C.; Alonso-Riaño, M.; Noguera-Morel, L.; Hernández-Martín, A.; Andina, D.; Wiesner, T.; Rodríguez-Peralto, J.L.; Requena, L.; Torrelo, A. SARS-CoV-2 endothelial infection causes COVID-19 chilblains: Histopathological, immunohistochemical and ultrastructural study of seven paediatric cases. Br. J. Dermatol. 2020, 183, 729–737.
- Fainardi, V.; Meoli, A.; Chiopris, G.; Motta, M.; Skenderaj, K.; Grandinetti, R.; Bergomi, A.; Antodaro, F.; Zona, S.; Esposito, S. Long COVID in Children and Adolescents. Life 2022, 12, 285.
- Li, W.T.; Zhang, Y.; Liu, M.; Liu, Y.Q.; Ma, X. Prolonged viral shedding in feces of children with COVID-19: A systematic review and synthesis of data. Eur. J. Pediatr. 2022, 181, 4011–4017.
- Santos, V.S.; Gurgel, R.Q.; Cuevas, L.E.; Martins-Filho, P.R. Prolonged Fecal Shedding of SARS-CoV-2 in Pediatric Patients: A Quantitative Evidence Synthesis. J. Pediatr. Gastroenterol. Nutr. 2020, 71, 150–152.
- Craddock, V.; Mahajan, A.; Spikes, L.; Krishnamachary, B.; Ram, A.K.; Kumar, A.; Chen, L.; Chalise, P.; Dhillon, N.K. Persistent circulation of soluble and extracellular vesicle-linked Spike protein in individuals with postacute sequelae of COVID-19. J. Med. Virol. 2023, 95, e28568.
- Dang, X.T.T.; Kavishka, J.M.; Zhang, D.X.; Pirisinu, M.; Le, M.T.N. Extracellular Vesicles as an Efficient and Versatile System for Drug Delivery. Cells 2020, 9, 2191.
- Flores-Alanis, A.; Sandner-Miranda, L.; Delgado, G.; Cravioto, A.; Morales-Espinosa, R. The receptor binding domain of SARS-CoV-2 spike protein is the result of an ancestral recombination between the bat-CoV RaTG13 and the pangolin-CoV MP789. BMC Res. Notes 2020, 13, 398.
- Scott, T.A.; Supramaniam, A.; Idris, A.; Cardoso, A.A.; Shrivastava, S.; Kelly, G.; Grepo, N.A.; Soemardy, C.; Ray, R.M.; McMillan, N.A.J.; et al. Engineered extracellular vesicles directed to the spike protein inhibit SARS-CoV-2. Mol. Ther. Methods Clin. Dev. 2022, 24, 355–366.
- Rotulo, G.A.; Palma, P. Understanding COVID-19 in children: Immune determinants and post-infection conditions. Pediatr. Res. 2023.
- Pou, C.; Nkulikiyimfura, D.; Henckel, E.; Olin, A.; Lakshmikanth, T.; Mikes, J.; Wang, J.; Chen, Y.; Bernhardsson, A.K.; Gustafsson, A.; et al. The repertoire of maternal anti-viral antibodies in human newborns. Nat. Med. 2019, 25, 591–596.
- Koch, C.M.; Prigge, A.D.; Anekalla, K.R.; Shukla, A.; Do Umehara, H.C.; Setar, L.; Chavez, J.; Abdala-Valencia, H.; Politanska, Y.; Markov, N.S.; et al. Age-related Differences in the Nasal Mucosal Immune Response to SARS-CoV-2. Am. J. Respir. Cell Mol. Biol. 2022, 66, 206–222.
- Brodin, P. SARS-CoV-2 infections in children: Understanding diverse outcomes. Immunity 2022, 55, 201–209.
- Varchetta, S.; Mele, D.; Oliviero, B.; Mantovani, S.; Ludovisi, S.; Cerino, A.; Bruno, R.; Castelli, A.; Mosconi, M.; Vecchia, M.; et al. Unique immunological profile in patients with COVID-19. Cell. Mol. Immunol. 2021, 18, 604–612.
- Abarca-Zabalía, J.; González-Jiménez, A.; Calle-Rubio, M.; López-Pastor, A.R.; Fariña, T.; Ramos-Acosta, C.; Anguita, E.; Urcelay, E.; Espino-Paisán, L. Alterations in the immune system persist after one year of convalescence in severe COVID-19 patients. Front. Immunol. 2023, 14, 1127352.
- Rojas, M.; Restrepo-Jiménez, P.; Monsalve, D.M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramírez-Santana, C.; Leung, P.S.C.; Ansari, A.A.; Gershwin, M.E.; Anaya, J.M. Molecular mimicry and autoimmunity. J. Autoimmun. 2018, 95, 100–123.
- Caso, F.; Costa, L.; Ruscitti, P.; Navarini, L.; Del Puente, A.; Giacomelli, R.; Scarpa, R. Could Sars-coronavirus-2 trigger autoimmune and/or autoinflammatory mechanisms in genetically predisposed subjects? Autoimmun. Rev. 2020, 19, 102524.
- Sundaresan, B.; Shirafkan, F.; Ripperger, K.; Rattay, K. The Role of Viral Infections in the Onset of Autoimmune Diseases. Viruses 2023, 15, 782.
- Di Sante, G.; Buonsenso, D.; De Rose, C.; Valentini, P.; Ria, F.; Sanguinetti, M.; Sali, M. Immune profile of children with post-acute sequelae of SARS-CoV-2 infection (Long COVID). medRxiv 2021.
- Gaebler, C.; Wang, Z.; Lorenzi, J.C.C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y.; et al. Evolution of antibody immunity to SARS-CoV-2. Nature 2021, 591, 639–644.
- Wang, E.Y.; Mao, T.; Klein, J.; Dai, Y.; Huck, J.D.; Jaycox, J.R.; Liu, F.; Zhou, T.; Israelow, B.; Wong, P.; et al. Diverse functional autoantibodies in patients with COVID-19. Nature 2021, 595, 283–288.
- Sun, J.; Xiao, J.; Sun, R.; Tang, X.; Liang, C.; Lin, H.; Zeng, L.; Hu, J.; Yuan, R.; Zhou, P.; et al. Prolonged Persistence of SARS-CoV-2 RNA in Body Fluids. Emerg. Infect. Dis. 2020, 26, 1834–1838.
- Zhou, M.; Yin, Z.; Xu, J.; Wang, S.; Liao, T.; Wang, K.; Li, Y.; Yang, F.; Wang, Z.; Yang, G.; et al. Inflammatory Profiles and Clinical Features of Coronavirus 2019 Survivors 3 Months After Discharge in Wuhan, China. J. Infect. Dis. 2021, 224, 1473–1488.
- Patterson, B.K.; Francisco, E.B.; Yogendra, R.; Long, E.; Pise, A.; Rodrigues, H.; Hall, E.; Herrera, M.; Parikh, P.; Guevara-Coto, J.; et al. Persistence of SARS CoV-2 S1 Protein in CD16+ Monocytes in Post-Acute Sequelae of COVID-19 (PASC) up to 15 Months Post-Infection. Front. Immunol. 2022, 12, 746021.
- Matsumiya, T.; Ota, K.; Imaizumi, T.; Yoshida, H.; Kimura, H.; Satoh, K. Characterization of synergistic induction of CX3CL1/fractalkine by TNF-alpha and IFN-gamma in vascular endothelial cells: An essential role for TNF-alpha in post-transcriptional regulation of CX3CL1. J. Immunol. 2010, 184, 4205–4214.
- Guilliams, M.; Mildner, A.; Yona, S. Developmental and Functional Heterogeneity of Monocytes. Immunity 2018, 49, 595–613.
- Kapellos, T.S.; Bonaguro, L.; Gemünd, I.; Reusch, N.; Saglam, A.; Hinkley, E.R.; Schultze, J.L. Human Monocyte Subsets and Phenotypes in Major Chronic Inflammatory Diseases. Front. Immunol. 2019, 10, 2035.
- Park, J.; Dean, L.S.; Jiyarom, B.; Gangcuangco, L.M.; Shah, P.; Awamura, T.; Ching, L.L.; Nerurkar, V.R.; Chow, D.C.; Igno, F.; et al. Elevated circulating monocytes and monocyte activation in COVID-19 convalescent individuals. Front. Immunol. 2023, 14, 1151780.
- Dowd, J.B.; Palermo, T.; Brite, J.; McDade, T.W.; Aiello, A. Seroprevalence of Epstein-Barr virus infection in U.S. children ages 6–19, 2003–2010. PLoS ONE 2013, 8, e64921.
- Neves, M.; Marinho-Dias, J.; Ribeiro, J.; Sousa, H. Epstein-Barr virus strains and variations: Geographic or disease-specific variants? J. Med. Virol. 2017, 89, 373–387.
- Gold, J.E.; Okyay, R.A.; Licht, W.E.; Hurley, D.J. Investigation of Long COVID Prevalence and Its Relationship to Epstein-Barr Virus Reactivation. Pathogens 2021, 10, 763.
- Houen, G.; Trier, N.H. Epstein-Barr Virus and Systemic Autoimmune Diseases. Front. Immunol. 2021, 11, 587380.
- Pallanti, S.; Di Ponzio, M. PANDAS/PANS in the COVID-19 Age: Autoimmunity and Epstein–Barr Virus Reactivation as Trigger Agents? Children 2023, 10, 648.
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793.
- Cipollaro, L.; Giordano, L.; Padulo, J.; Oliva, F.; Maffulli, N. Musculoskeletal symptoms in SARS-CoV-2 (COVID-19) patients. J. Orthop. Surg. Res. 2020, 15, 178.
- Beydon, M.; Chevalier, K.; Al Tabaa, O.; Hamroun, S.; Delettre, A.S.; Thomas, M.; Herrou, J.; Riviere, E.; Mariette, X. Myositis as a manifestation of SARS-CoV-2. Ann. Rheum. Dis. 2021, 80, e42.
- Manzano, G.S.; Woods, J.K.; Amato, A.A. Covid-19-Associated Myopathy Caused by Type I Interferonopathy. N. Engl. J. Med. 2020, 383, 2389–2390.
- Movahedi, N.; Ziaee, V. COVID-19 and myositis; true dermatomyositis or prolonged post viral myositis? Pediatr. Rheumatol. 2021, 19, 86.
- Tanboon, J.; Nishino, I. COVID-19-associated myositis may be dermatomyositis. Muscle Nerve 2021, 63, E9–E10.
- Qian, J.; Xu, H. COVID-19 Disease and Dermatomyositis: A Mini-Review. Front. Immunol. 2022, 12, 747116.
- Gokhale, Y.; Patankar, A.; Holla, U.; Shilke, M.; Kalekar, L.; Karnik, N.D.; Bidichandani, K.; Baveja, S.; Joshi, A. Dermatomyositis during COVID-19 Pandemic (A Case Series): Is there a Cause Effect Relationship? J. Assoc. Physicians India 2020, 68, 20–24.
- Crivelenti, L.R.M.P.; Frazão, M.M.N.; Maia, M.P.M.; Gomes, F.H.R.; de Carvalho, L.M. Chronic arthritis related to SARS-CoV-2 infection in a pediatric patient: A case report. Braz. J. Infect. Dis. 2021, 25, 101585.
- Sinaei, R.; Pezeshki, S.; Parvaresh, S.; Sinaei, R.; Shiari, R.; Hassas Yeganeh, M.; Bazargn, N.; Gharaei, N. Post SARS-CoV-2 infection reactive arthritis: A brief report of two pediatric cases. Pediatr. Rheumatol. Online J. 2021, 19, 89.
- Ailioaie, L.M.; Ailioaie, C.; Litscher, G. Implications of SARS-CoV-2 Infection in Systemic Juvenile Idiopathic Arthritis. Int. J. Mol. Sci. 2022, 23, 4268.
- Zitouni, J.; Bursztejn, A.C.; Belloni Fortina, A.; Beauchet, A.; Di Lernia, V.; Lesiak, A.; Thomas, J.; Topkarci, Z.; Murashkin, N.; Brzezinski, P.; et al. Children with psoriasis and COVID-19: Factors associated with an unfavourable COVID-19 course, and the impact of infection on disease progression (Chi-PsoCov registry). J. Eur. Acad. Dermatol. Venereol. 2022, 36, 2076–2086.
- Alexander, A.J.; Joshi, A.; Mehendale, A. The Musculoskeletal Manifestations of COVID-19: A Narrative Review Article. Cureus 2022, 14, e29076.
- Pal, A.; Roongta, R.; Mondal, S.; Sinha, D.; Sinhamahapatra, P.; Ghosh, A.; Chattopadhyay, A. Does post-COVID reactive arthritis exist? Experience of a tertiary care centre with a review of the literature. Reumatol. Clin. 2023, 19, 67–73.
- Kumar, P.; Jat, K.R. Post-COVID-19 Sequelae in Children. Indian J. Pediatr. 2023, 90, 605–611.
- Ailioaie, L.M.; Ailioaie, C.; Litscher, G.; Chiran, D.A. Celiac Disease and Targeting the Molecular Mechanisms of Autoimmunity in COVID Pandemic. Int. J. Mol. Sci. 2022, 23, 7719.
- Chen, B.; Julg, B.; Mohandas, S.; Bradfute, S.B. RECOVER Mechanistic Pathways Task Force (2023). Viral persistence, reactivation, and mechanisms of long COVID. Elife 2023, 12, e86015.
- de Oliveira, G.L.V.; Oliveira, C.N.S.; Pinzan, C.F.; de Salis, L.V.V.; Cardoso, C.R.B. Microbiota Modulation of the Gut-Lung Axis in COVID-19. Front. Immunol. 2021, 12, 635471.
- Troisi, J.; Venutolo, G.; Pujolassos Tanyà, M.; Delli Carri, M.; Landolfi, A.; Fasano, A. COVID-19 and the gastrointestinal tract: Source of infection or merely a target of the inflammatory process following SARS-CoV-2 infection? World J. Gastroenterol. 2021, 27, 1406–1418.
- Settanni, C.R.; Ianiro, G.; Ponziani, F.R.; Bibbò, S.; Segal, J.P.; Cammarota, G.; Gasbarrini, A. COVID-19 as a trigger of irritable bowel syndrome: A review of potential mechanisms. World J. Gastroenterol. 2021, 27, 7433–7445.
- Dhar, D.; Mohanty, A. Gut microbiota and Covid-19- possible link and implications. Virus Res. 2020, 285, 198018.
- Azer, S.A. COVID-19: Pathophysiology, diagnosis, complications and investigational therapeutics. New Microbes New Infect. 2020, 37, 100738.
- Chakaroun, R.M.; Massier, L.; Kovacs, P. Gut Microbiome, Intestinal Permeability, and Tissue Bacteria in Metabolic Disease: Perpetrators or Bystanders? Nutrients 2020, 12, 1082.
- Tsounis, E.P.; Triantos, C.; Konstantakis, C.; Marangos, M.; Assimakopoulos, S.F. Intestinal barrier dysfunction as a key driver of severe COVID-19. World J. Virol. 2023, 12, 68–90.
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379.
- Kwak, B.O.; Eun, B.W. COVID-19 in immunocompromised children and adolescents. Clin. Exp. Pediatr. 2023, 66, 182–189.
- Acosta-Ampudia, Y.; Monsalve, D.M.; Rojas, M.; Rodríguez, Y.; Zapata, E.; Ramírez-Santana, C.; Anaya, J.M. Persistent Autoimmune Activation and Proinflammatory State in Post-Coronavirus Disease 2019 Syndrome. J. Infect. Dis. 2022, 225, 2155–2162.
- Queiroz, M.A.F.; Neves, P.F.M.D.; Lima, S.S.; Lopes, J.D.C.; Torres, M.K.D.S.; Vallinoto, I.M.V.C.; Bichara, C.D.A.; Dos Santos, E.F.; de Brito, M.T.F.M.; da Silva, A.L.S.; et al. Cytokine Profiles Associated with Acute COVID-19 and Long COVID-19 Syndrome. Front. Cell. Infect. Microbiol. 2022, 12, 922422.
- Yin, J.X.; Agbana, Y.L.; Sun, Z.S.; Fei, S.W.; Zhao, H.Q.; Zhou, X.N.; Chen, J.H.; Kassegne, K. Increased interleukin-6 is associated with long COVID-19: A systematic review and meta-analysis. Infect. Dis. Poverty 2023, 12, 43.
- Al-Sadi, R.; Dharmaprakash, V.; Nighot, P.; Guo, S.; Nighot, M.; Do, T.; Ma, T.Y. Bifidobacterium bifidum Enhances the Intestinal Epithelial Tight Junction Barrier and Protects against Intestinal Inflammation by Targeting the Toll-like Receptor-2 Pathway in an NF-κB-Independent Manner. Int. J. Mol. Sci. 2021, 22, 8070.
- Ailioaie, L.M.; Litscher, G. Probiotics, Photobiomodulation, and Disease Management: Controversies and Challenges. Int. J. Mol. Sci. 2021, 22, 4942.
- Bacorn, M.; Romero-Soto, H.N.; Levy, S.; Chen, Q.; Hourigan, S.K. The Gut Microbiome of Children during the COVID-19 Pandemic. Microorganisms 2022, 10, 2460.
- Yonker, L.M.; Gilboa, T.; Ogata, A.F.; Senussi, Y.; Lazarovits, R.; Boribong, B.P.; Bartsch, Y.C.; Loiselle, M.; Rivas, M.N.; Porritt, R.A.; et al. Multisystem inflammatory syndrome in children is driven by zonulin-dependent loss of gut mucosal barrier. J. Clin. Investig. 2021, 131, e149633.
- Yeoh, Y.K.; Zuo, T.; Lui, G.C.; Zhang, F.; Liu, Q.; Li, A.Y.; Chung, A.C.; Cheung, C.P.; Tso, E.Y.; Fung, K.S.; et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 2021, 70, 698–706.
- Haran, J.P.; Bradley, E.; Zeamer, A.L.; Cincotta, L.; Salive, M.C.; Dutta, P.; Mutaawe, S.; Anya, O.; Meza-Segura, M.; Moormann, A.M.; et al. Inflammation-type dysbiosis of the oral microbiome associates with the duration of COVID-19 symptoms and long COVID. JCI Insight 2021, 6, e152346.
- Ancona, G.; Alagna, L.; Alteri, C.; Palomba, E.; Tonizzo, A.; Pastena, A.; Muscatello, A.; Gori, A.; Bandera, A. Gut and airway microbiota dysbiosis and their role in COVID-19 and long-COVID. Front. Immunol. 2023, 14, 1080043.
- Gareau, M.G.; Barrett, K.E. Role of the microbiota-gut-brain axis in postacute COVID syndrome. Am. J. Physiol. Gastrointest. Liver Physiol. 2023, 324, G322–G328.
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013.
- Bostick, J.W.; Schonhoff, A.M.; Mazmanian, S.K. Gut microbiome-mediated regulation of neuroinflammation. Curr. Opin. Immunol. 2022, 76, 102177.
- Ailioaie, L.M.; Ailioaie, C.; Litscher, G. Photobiomodulation in Alzheimer’s Disease—A Complementary Method to State-of-the-Art Pharmaceutical Formulations and Nanomedicine? Pharmaceutics 2023, 15, 916.
- Lorens, S.; Nava, E.; Muñoz-López, M.; Sánchez-Larsen, Á.; Segura, T. Neurological Symptoms of COVID-19: The Zonulin Hypothesis. Front. Immunol. 2021, 12, 665300.
- Okuyucu, M.; Yalcin Kehribar, D.; Çapraz, M.; Çapraz, A.; Arslan, M.; Çelik, Z.B.; Usta, B.; Birinci, A.; Ozgen, M. The Relationship Between COVID-19 Disease Severity and Zonulin Levels. Cureus 2022, 14, e28255.
- Al Bataineh, M.T.; Henschel, A.; Mousa, M.; Daou, M.; Waasia, F.; Kannout, H.; Khalili, M.; Kayasseh, M.A.; Alkhajeh, A.; Uddin, M.; et al. Gut Microbiota Interplay With COVID-19 Reveals Links to Host Lipid Metabolism Among Middle Eastern Populations. Front. Microbiol. 2021, 12, 761067.
- Reinold, J.; Farahpour, F.; Fehring, C.; Dolff, S.; Konik, M.; Korth, J.; van Baal, L.; Hoffmann, D.; Buer, J.; Witzke, O.; et al. A Pro-Inflammatory Gut Microbiome Characterizes SARS-CoV-2 Infected Patients and a Reduction in the Connectivity of an Anti-Inflammatory Bacterial Network Associates with Severe COVID-19. Front. Cell. Infect. Microbiol. 2021, 11, 747816.
- Cheng, X.; Zhang, Y.; Li, Y.; Wu, Q.; Wu, J.; Park, S.K.; Guo, C.; Lu, J. Meta-analysis of 16S rRNA microbial data identified alterations of the gut microbiota in COVID-19 patients during the acute and recovery phases. BMC Microbiol. 2022, 22, 274.
- Guido, C.A.; Lucidi, F.; Midulla, F.; Zicari, A.M.; Bove, E.; Avenoso, F.; Amedeo, I.; Mancino, E.; Nenna, R.; De Castro, G.; et al. Long-Covid Group of Department of Maternal Sciences. Neurological and psychological effects of long COVID in a young population: A cross-sectional study. Front. Neurol. 2022, 13, 925144.
- Schneider, K.M.; Blank, N.; Alvarez, Y.; Thum, K.; Lundgren, P.; Litichevskiy, L.; Sleeman, M.; Bahnsen, K.; Kim, J.; Kardo, S.; et al. The enteric nervous system relays psychological stress to intestinal inflammation. Cell 2023, 186, 2823–2838.e20.
- Sidik, S. Chronic stress can inflame the gut-now scientists know why. Nature 2023, 618, 221–222.