Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Immunology
Virus-derived nucleic acids are potential immune-enhancers and particularly good candidates as adjuvants in vaccines in mouse models. The most important nucleic-acid-sensing process involves the dendritic cell (DC) Toll-like receptor (TLR), which participates in the pattern recognition of foreign DNA/RNA structures. Human CD141+ DCs preferentially express TLR3 in endosomes and recognize double-stranded RNA. Antigen cross-presentation occurs preferentially in this subset of DCs (cDCs) via the TLR3–TICAM-1–IRF3 axis. Another subset, plasmacytoid DCs (pDCs), specifically expresses TLR7/9 in endosomes.
- Toll-like receptor 3
- dendritic cell
- Th1 polarization
- cross-antigen presentation
1. Introduction
Humans sustain their survival via constant exposure to diverse microbial environments. Human populations exhibit heterogeneous reactivity to infections from individual to individual via environmental selection. The host response, rather than the microorganisms themselves, is the main driver of infectious diseases. Vaccines, on the other hand, have been discussed empirically in the context of the “no-twice-infected phenomenon” and “post-Jenner vaccines”. It can be said that the immune response has been established as a mechanism for eliminating microorganisms, but the immune response to vaccines varies from individual to individual in each infectious disease and also to mutant strains. In addition, both excessive responses and adverse reactions induce inappropriate immune aberrance in the host.
Vaccines are being developed primarily as therapeutic and prophylactic vaccines for cancer and infectious diseases. In both cases, cDC priming with safe adjuvants has been proven to be essential. However, non-toxic and versatile adjuvants that can be used in the elderly and patients with underlying diseases have not been established.
Adjuvants have been classified as receptor-defined and undefined. Alum (aluminum salt) has been widely used in licensed vaccines targeting pathogens for humans, but the receptor is still undefined [1]. Saponins regulate the Th1/2 polarization of DCs, but their direct receptors are undefined [1]. Alum vaccines have the advantage of increasing antibody production under Th2 situation, but the causal attribution of adjuvant activity is unknown in alum, implying that no method exists to rescue it from an adverse event in case it occurs. In contrast, nucleic acid adjuvants have defined receptors, such as TLRs, and TLR signaling has been clarified at the molecular level.
Subcomponent vaccines usually consist of an antigen and an adjuvant. An adjuvant is a substance that enhances the immune response against non-self antigens in vertebrates, including humans, and is an essential component of vaccines [1]. Adjuvants (i) modulate the rate, time duration, and quality of the immune response; (ii) maximize the immune response to vaccine antigens; and (iii) induce antigen-specific acquired immunity [2]. Multiple mechanisms exist in the action of adjuvants in a broad sense. Although the molecular mechanism for (i) is not always well understood, it promotes sustained stimulation of the immune system via depot formation, in which the antigen is retained and slowly released [1,3,4]; (ii) and (iii) refer to a narrow-sense adjuvant that can be functionally defined as a ligand–receptor molecular response. It is crucial for adjuvants to promote the recruitment of immune cells and facilitate the maturation of antigen-presenting dendritic cells (cDCs) via the production of cytokines and chemokines [1,3].
Once mature, cDCs acquire the ability to process and cross-present extrinsic antigens on MHC class I molecules and induce a specific immune response against foreign non-self antigens via the proliferation of lymphocytes [1,4]. In this context, adjuvants can modulate the activation mode of DCs to induce a variety of CD4+ T cell subsets [4]. Th1 polarization creates a favorable environment for the induction of cellular and humoral immunity. Th2 polarization strongly activates humoral immunity but suppresses cellular immunity. The Th17 shift contributes to defense against infections, and the Th22 shift induces a more specific immune response [4,5]. Th17 and Th22 cells (releasing interleukin (IL)-17 and IL-22, respectively) especially play important roles in defending against fungal and bacterial infections on mucosal surfaces, including the lungs and gut. These are in part attributed to the fact that adjuvants induce different types of cytokines/chemokines [6]. Selecting an appropriate adjuvant for the target antigen (from the pathogen) is indispensable for a successful vaccination.
Several other functions appear, besides cDC maturation, in antitumor nucleic acid adjuvants. They can act on stroma cells to reprogram the tumor microenvironment (TME) [6,7]. Cytotoxic T lymphocyte (CTL)-attracting chemokines are released in response to RNA adjuvants. However, what happens to adjuvants in the pathogen-associated microenvironment during vaccination remains unsolved.
2. Alum Adjuvants
Alum adjuvants have been widely used in vaccines against human infectious diseases. Alum adjuvants, when combined with antigens, potentially primarily induce a Th2 response and antibody production, and thus have been used in many routine and voluntary vaccines that successfully protect against pathogens via antibody responses [1,4]. Alum has been an appropriate and safe component of licensed vaccines against pathogens for approximately 80 years. The possible mechanisms for the immune-potentiating effects of alum include antigen depot properties, in which antigens accumulate and are slowly released from immune sites [3,4,8]. Skin irritation and fever caused by alum adjuvants have been reported to be mild, but the mechanism of action of the alum adjuvants remains unknown. Furthermore, alum adjuvants may induce the excessive activation of Th2-polarized immunity, leading to adverse effects such as the promotion of autoimmune diseases [9] and antibody- or vaccine-dependent disease enhancement (ADE/VDE) [10,11]. Moreover, the Th2 shift in acquired immunity promotes tumor cell proliferation and lifestyle diseases [12,13]. On the contrary, alum is ineffective against infections such as severe acute respiratory syndrome (COVID-19) and respiratory syncytial virus infections, where T cell immunity is essential for the early elimination of the pathogen [11,14]. Therefore, a new adjuvant is desired that covers both the antigen-specific antibody and cellular responses in Th1 polarization without toxicity.
In this context, researchers explored the possibility of adapting nucleic acid adjuvants currently used for cancer to be used as vaccines against infectious diseases. These adjuvants act on Toll-like receptors (TLRs) in DCs, and preferentially induce Th1 polarization, referred to as Th1 adjuvants. As the receptors for DNA/RNA in DCs are identified and common, in principle, across humans and mice, the mechanism of DC maturation can be analyzed in mouse models. The function of DC-mediated Th1 skewing can be molecularly defined, laying out the results for mice in humans. The role of Th1 adjuvants in vaccines is, to researchers understanding, in association with DC maturation and cross-antigen presentation.
3. DCs and TLRs
DCs are short-lived (3−5 days) bone-marrow-derived cells that may be supplied locally from the blood pool at any time, differentiating from myeloid precursors to macrophage–DC progenitors and then to common DC progenitors (CDPs) in bone marrow [15]. CDPs are differentiated into DCs by Flt3L [15,16]. Pre-DCs then migrate from the bone marrow into the blood and differentiate into conventional DCs (cDCs) in secondary lymphoid tissues and other tissues. In contrast, the cells differentiated from CDPs to plasmacytoid DCs (pDCs) in the bone marrow enter the blood and migrate to the tissues [15].
The following three types of signals are thought to be simultaneously initiated in activating DCs associated with infection [17,18]. The first comprises inflammatory cytokines such as tumor necrosis factor α released from myeloid cells that infiltrate the infected area [17], the second comprises components derived from dead cells such as neutrophils and macrophages that die as a result of infection (i.e., DAMPs) [19,20], and the third comprises signals from TLRs that recognize components derived from bacteria and viruses (e.g., lipopolysaccharides from Gram-negative bacteria and dsRNA from viruses) [21]. The third signal from TLRs participates in the specific targeting of cDCs, evoking the acquired immune system to eradicate infections [21,22]. The cDCs remain at the site of infection for several hours, take up sufficient antigens and become activated, and then migrate through the lymph vessels to their lymph nodes (LNs), where they activate naive T cells, ending their life span within 1 week [18,23]. The infected lesion is then supplied with new DCs from the bone marrow, and as long as the infection continues, the steps of activation of DCs at the site of infection and migration to the regional lymph nodes are repeated periodically [23]. The life cycle and behavior of DCs completely differ from those of macrophages, which stay in the infectious area. Via migration to local LNs, antigen-presenting DCs specify their function to the cross-antigen presentation of foreign antigens through their maturation [4].
CD141+ DCs are professional DC subsets for cross-antigen presentation in humans. This DC subsets correspond to mouse DC subsets CD8α+ and CD103+ [18] and commonly express XCR1 [24]. Thus, XCR1+ is a representative marker reflecting the function of cross-antigen presentation in DCs. CD141+ DCs are BDCA3-positive, which represents the expression of thrombomodulin protein on the cell-surface [24].
CD141+ DCs express TLR3 in endosomes, and are upregulated in response to IFN-I [18]. Recent studies have shown that TLR3 in cDCs can be targeted for activation via a synthetic ligand ARNAX or Riboxxim (Figure 1) [25,26]. In contrast, CD1c+ DCs express TLR4 on the cell surface, and are activated in response to lipopolysaccharide (LPS) via two arrays of adaptors, MyD88 and TICAM-1 (TRIF) [27,28]. Lipid A is an active center for TLR4. The structure of Lipid A determines the biased activation of either MyD88 or TICAM-1 [29,30]. Previous studies have suggested that monophosphoryl Lipid A (MPLA) predominantly activates the TICAM-1 pathway, allowing it to accomplish a less toxic TLR4 agonist [29,31,32]. Unlike TLR3, TLR4 requires TICAM-2 (TRAM) to link TICAM-1 [30]. TLR4 and TLR3 share the TICAM-1 pathway that leads to IL-12 secretion and Th1 skewing [28,29], and contribute to safe DC priming [31,32,33]. IFN-γ plays a pivotal role in establishing the Th1 shift [32]. MPLA has been used for preparing several preventive vaccines against infectious diseases [34,35]. TLR4 may evoke antiviral immunity [36], but does not recognize nucleic acids.
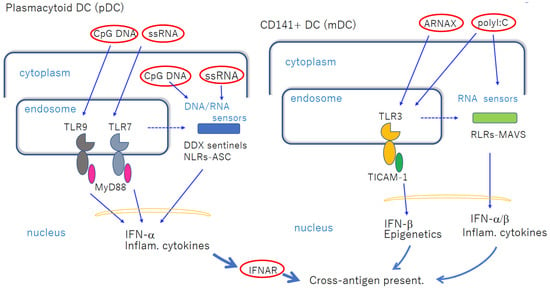
Figure 1. Nucleic-acid-sensing TLRs in pDCs and cDCs facilitate cross-antigen presentation. Left panel: pDCs express TLR7 and TLR9 in endosomes, which recognize imidazoquinoline and CpG DNA, respectively, when exogenously added. MyD88 transduces the signal to NF-κB activation to induce robust inflammatory cytokines and IFN-α. These mediators act on cDCs to induce cross-antigen presentation. If the ligands are transfected into cytoplasm, they are recognized using cytoplasmic sensors. pDCs do not present antigens per se. Right panel: CD141+ DCs express TLR3, and are increased via stimulation with dsRNA. Although blunt-ended viral dsRNA barely enters the endosome, ARNAX and PLGA-Riboxxim enter the endosome to activate TLR3 without transfection reagent. polyI:C activates both TLR3 and RLRs without transfection. Break lines indicate putative routes of nucleic acids to move from endosome to cytoplasm. The availability of this route depends on the cell types and properties of ligands. The properties of the transfection reagent affect the delivery of DNA/RNA. cDCs mature via direct TLR3-targeting without significant cytokine liberation in circulation.
Both human and mouse antigen-presenting DCs are XCR1-positive, and express TLR3 in common and respond to ARNAX. However, mouse antigen-presenting DCs, including CD8α+ and CD103+ subsets, express TLR4, while their human counterparts CD141+ DCs do not [18].
4. pDCs and cDCs in Recognition of Nucleic Acid Adjuvants
The human pDC subset possesses the MyD88 pathway for TLR9/7-ligand-stimulated activation [37,38]. pDCs express TLR9/7 in endosomes and recognize the unmethylated CpG DNA and single-stranded RNA of viral species to produce IFN-I [37,38]. The transportation of the TLR complexes into the endosomal compartment requires the adapter AP3 [39]. Rather than viral DNA/RNA, synthetic ligands (such as oligonucleotides (ODNs) and quinoline-derivatives) have been used to elucidate TLR9/7-specific signaling pathways [40] (Figure 1). pDC activation results in the production of robust IFN and inflammatory cytokines, and these cytokines directly eliminate viruses [37,38]. pDCs lack the ability to present antigens [40]. K-type ODNs directly stimulate pDCs and B cells and promote antibody production [41]. TLR9 favors B-cell-mediated antibody production in comparison with TLR3. CpG ODN (but not GpC ODN) stimulation in B cells promotes preB cell differentiation into plasma cells and memory B cells. PreproB (naive B) alters CD5+ B cells to produce IL-10 and suppresses IL-12 production by cDCs, hindering Th1 polarity [42]. Furthermore, the robust cytokine storm caused by pDC activation can exacerbate smoldering inflammation and autoimmune disorders.
pDCs release IFN-I/cytokines and secondarily activate antigen-presenting DCs in the local environment. pDCs enhance cDC cross-antigen presentation via released IFN-I/cytokines that are rooted in pDC nucleosensing (Figure 1). Inflammation is an absolute prerequisite for such cDC-driven cross-antigen presentation. Thus, pDCs participate in this “inflammatory mode” of antigen presentation via cDC maturation. As severe inflammation can lead to Th2 polarization, Th1 suppression, and tumor growth [43], ligand formulations have been devised to reduce these side effects in nucleic acid adjuvants. pDC-specific targeting by TLR9/7 agonists is worth developing, but it must be improved in clinical trials without exacerbating diseases.
In addition, human DCs, including pDCs, respond to R848, a common ligand for TLR7 and TLR8 [44]. Both TLR7 and 8 appear to be activated in response to single-stranded RNAs with different AU/GC content [44]. VTX-2337 is known to specifically trigger TLR8 activation. It is known that TLR8 stimulation promotes an NK-cell-dependent immune response against tumors, which is accompanied by increased IFN-γ production [44,45]. Thus, direct action of NK cells by TLR8 ligands promotes tumor regression [46]. Human-bone-marrow-derived suppressor cells express TLR8. Human DCs, macrophages, and monocytes also express TLR8, and human immature myeloid cells express TLR8 at high levels; the role of DC-specific TLR8 is not clearly demonstrated by the addition of TLR8 agonists to cultured human cells. In addition, it is difficult to obtain results with TLR8 agonists in mice.
In contrast, the human cDC subset CD141+ DCs predominantly express TLR3 [47], which recognizes dsRNA and transmits signals via the TICAM-1 adapter to mature cDCs for cross-antigen presentation [48]. This is “a non-inflammatory mode” for direct DC priming. Except for direct cDC infection (such as by the measles virus) [49], viral blunted dsRNAs derived from infected cells barely enter the endosomes of DCs [50,51]. Naked viral dsRNAs thereby induce no DC maturation [50,51]. The belief at present is that exosomes containing dsRNAs may deliver viral dsRNAs to cDC endosomes [52]. However, multiple DNA/RNA sensors localized in the cytoplasm, especially known as the DEAD-box helicase family (or sentinels), may co-work with TLR3 [52,53]. Hence, vaccination and actual infection differ in the aforementioned terms.
Viral dsRNA entering from the outside is mostly AU-rich and rarely reaches the endosomes where TLR3 is localized [25,51]. For the extrinsic use of dsRNAs as DC-targeting adjuvants, it is essential to appropriately design dsRNAs to guide them to the endosomes [25,26]. Several studies have demonstrated that the modification of dsRNAs facilitated them to gain access to endosomal TLR3 [25,26] (Table 1). ARNAX is 5′-GpC DNA-capped AU-rich dsRNA (120~140 bp) of a measles leader–trailer sequence, while PLGA-Riboxxim consists of PLGA and 100 bp GC-rich dsRNA (Table 1). In contrast, recent studies have suggested that ~424-bp dsRNA generated via in vitro transcription had TLR3-agonistic activity without transfection, which was similar to that of polyI:C [2,49]. The nucleotide segment was from the Chinese Sacbrood virus genome [54], implying that the nucleotide sequence would be a determinant in dsRNAs for internalization into DCs.
Table 1. Structure-defined dsRNA.
| Items | ARNAX | PLGA-Riboxxim | NexaVant |
|---|---|---|---|
| dsRNA size(bp) | 120~140 | 100 | 424 |
| Structurally defined | yes | yes | yes |
| endosomal delivery | 5′-DNA | PLGA adsorbed | Internal sequence |
| Target receptor | TLR3 | TLR3/RIG-I | TLR3 |
| Target cells | CD104+DC | CD104+, CD1c+DC | DC? |
| pathway | TLR3 | TLR3/MAVS | TLR3/MAVS? |
| stability | high | high | high |
| GMP possibility | yes | yes | yes |
| Date of first publication | 2015 | 2021 | 2023 |
Hence, nucleic acids with appropriate formulation can enter endosomes with TLR3, making it possible to sense nucleic acids via TLR3 [2,25,26]. This non-inflammatory mode concerns the direct DC-priming mode [4,55]. In either mode, antigen-presenting DCs play a pivotal role in evoking the acquired immune system, where antigen-specific T cells proliferate.
This entry is adapted from the peer-reviewed paper 10.3390/cells12111504
This entry is offline, you can click here to edit this entry!