The term α-synucleinopathies defines a group of neurodegenerative disorders associated with pathological accumulation of α-Synuclein (αSyn) aggregates in neurons and non-neuronal cells including microglia, pericytes, astrocytes, and oligodendrocytes [
1]. Clinically, α-synucleinopathies comprise Lewy body disease (LBD) and multiple system atrophy (MSA). LBD is associated with abnormal accumulation of insoluble aggregated αSyn in Lewy bodies (LBs) and Lewy neurites (LNs) of neurons [
2] such as in Parkinson’s disease (PD), PD dementia (PDD), dementia with Lewy bodies (DLB), and other neurodegenerative disorders [
3]. MSA is a different synucleinopathy which includes two major clinicopathological subtypes characterized by the presence of argyrophilic glial cytoplasmic inclusions (GCI) and neuronal loss accompanied by gliosis in the basal ganglia, cerebellum, pons, inferior olivary nuclei, and spinal cord [
4]. GCI have been reported also to consist of αSyn; therefore, MSA and LBD are considered the two major subtypes of synucleinopathies [
5].
αSyn is a small protein composed by 140 amino acids belonging to the synucleins family (α-synuclein, β-synuclein, γ-synuclein, and synoretin), whose members share high sequence identity and expression pattern [
9]. Synucleins are natively unfolded proteins characterized by an acidic carboxyl terminus and an amino terminus containing imperfect repeat motifs (KTKEGV) [
10]. Human αSyn is predominantly expressed in the brain and highly abundant in the presynaptic terminal of dopaminergic neurons. Phosphorylation and dephosphorylation of residue of serine 129 (pS129-αSyn) are known to be responsible for promoting or inhibiting the α-Syn aggregation, respectively [
11,
12,
13]. However, the pathogenic relevance of pS129-αSyn remains controversial, as a recent finding shows that pS129-αSyn inhibits fibril formation and seeded aggregation [
12]. It has been reported that the truncation of C-terminus of recombinant αSyn promotes its assembly to potentially form pathological filaments [
14,
15].
Predominantly cytosolic and partly nuclear, αSyn can also be localized at the level of lipid membranes. The presence of αSyn was also found at the level of the mitochondrial membranes, internal and external, where it interferes with the activity of complex I and IV of the respiratory chain. Impairment of these complexes can lead to increased ROS production, often a cause of neuronal death [
20,
21,
22]. αSyn is able to inhibit phospholipase D2, a membrane enzyme involved in the release of phosphatidic acid, useful for the formation of membranous vesicles and synaptic membranes [
23,
24,
25] and to interact with different synaptic proteins by modulating their activity. In particular, its interaction with small GTPases (Rab) has been shown to be important in modulating membrane trafficking, exocytosis and synaptic vesicle release [
26,
27,
28,
29]. It is important to underline that the protein-protein interactions at the cytosolic level can influence the physiological and biochemical functions of αSyn and consequently its tendency to aggregate. This suggests that it is possible to modify the aggregation of αSyn by selectively regulating the expression of its protein partners [
30,
31].
2. Protein Aggregation and Propagation in Synucleinopathies
The proteostasis network works to maintain the proper balance between protein synthesis, folding, and degradation. It includes the ubiquitin-proteasome system, autophagy, chaperons, and heat shock proteins. Together, these pathways are responsible for regulating the number and the quality of proteins in the cell as well as their clearance.
Perturbations in proteostasis have been associated with the pathogenesis of synucleinopathies [
6]. αSyn is an intrinsically disordered protein that does not have a single, well-defined three-dimensional structure, but exists in a dynamic equilibrium of multiple conformations. While the normal expression of αSyn has a role in protecting cells against oxidative stress and apoptosis, its accumulation and the consequent formation of particular states of aggregation, such as oligomers or fibrils, may be harmful to the cell. It has been demonstrated that in certain conditions, such as oxidative stress, nitrosative stress, and inflammation, αSyn can misfold into amyloid-like fibrils. Once misfolded, αSyn can form cell-to-cell aggregates which can spread throughout the brain and cause further damage [
6,
35]. This hypothesis has been further supported by other studies, which have found that αSyn expression levels can influence the progression of diseases such as PD [
36], Alzheimer disease (AD) [
37], and Huntington’s disease (HD) [
38]. Although it is still debated whether αSyn aggregation is the main leading cause for the neuronal death, to date, oligomeric/aggregated species of αSyn contained in the LBs and LNs are considered the hallmarks of PD and other synucleinopathies [
39,
40,
41]. There is a different susceptibility of specific brain areas to LB and LN formation. For example, due to the presence of numerous neurites and terminations and the high requirement for mitochondrial activity, dopaminergic neurons represent the cells most vulnerable to LB and LN build-up [
42,
43,
44]; LBs and LNs are preferentially formed in nigrostriatal neurons with long-range projections [
45,
46,
47]. Hyperbranching axons as in the nigrostriatal system may facilitate αSyn deposition [
48]. These neurons are more vulnerable to oxidative and nitrosative stress [
49,
50,
51,
52]. It has been proposed that the progression of Lewy pathology is from axon terminals to neuronal soma. However, a “mitocentric view of PD” claims that mitochondrial dysfunctions can lead to nigrostriatal degeneration regardless of Lewy pathology [
48].
The exact mechanism of αSyn misfolding is still not fully understood, even if it is thought to involve a combination of environmental and genetic factors [
53,
54,
55]. Studies have suggested that misfolding may be caused by exposure to toxins, age-related alterations in the αSyn structure, and gene mutations, and may cause an increased propensity to the formations of αSyn aggregates and fibrils [
42,
55].
αSyn aggregation initiates by a short sequence of a seed or native, partially folded or unfolded oligomers, which adopt a non-native conformation and auto-assemble into higher-order oligomers. These oligomers can serve as precursors of fibril nucleus, highly dynamic species that recruit other monomers eliciting a rapid polymerization into amyloid fibrils, hierarchical polymorphic stable structures derived from protofibrils that are responsible for the development of several diseases. The current goal is to identify the exact intermediate structures and pathways involved in certain diseases and that are characteristic for each individual. Since amyloid fibril-associated diseases are orphan drug, building a molecular profile for a group of individuals with certain feature clinical signs could improve the diagnosis and be helpful for the designing of molecules able to potentially prevent the pathological escalation.
3. Toll-like Receptors (TLRs) in α-Synuclein Aggregation
The Toll-like receptors (TLRs) represent a family (at least 10 members TLR1-TLR10) of transmembrane proteins expressed by immune and non-immune cells, including microglia, neurons, astrocytes, and oligodendrocytes involved in the activation of the innate immune system [
70]. They are located on the surface of cells and recognize pathogen-associated molecular patterns (PAMPs) derived from bacteria, viruses, fungi, and other pathogens or can also be found in the endosome and cytoplasm where they detect and respond to viral nucleic acids [
71,
72]. TLRs are also able to recognize a wide variety of damage- or danger-associated molecular patterns (DAMPs), also known as alarmins, including αSyn, released by damaged neuronal cells and injured tissues [
73]. Upon recognition of these molecules, TLRs activate signaling pathways that lead to the production of pro-inflammatory cytokines, and can recruit and activate other immune cells, such as T cells and B cells initiating an adaptive immune response [
74,
75,
76].
Downstream signaling pathways of TLRs include the myeloid differentiation primary-response gene 88 (MyD88), MyD88-adaptor-like protein (MAL), TIR-domain-containing adaptor protein inducing interferon-β (IFNβ) (TRIF), TRIF-related adaptor molecule (TRAM), and sterile α- and armadillo-motif-containing protein (SARM). Except for TLR3, MyD88 is a key component of the TLR signaling pathway. It is a cytoplasmic adaptor protein that, upon recruitment, activates signaling molecules such as IL-1R-associated (IRAK) kinases, TNF receptor-associated factors (TRAFs), and TAK1 protein kinase complex, starting a signaling cascade that in turn leads to the activation of NF-kB and finally to the production of pro-inflammatory cytokines [
77].
TLRs and αSyn are shown to be reciprocally influenced in a positive feedback loop; indeed, αSyn increases the expression of TLRs, including TLR1, TLR2, TLR3, and the adaptor Myd88 [
77] and TLR2 and TLR4 are dysregulated in PD patients and animal models [
78,
79,
80,
81]. TLR dysregulation has been linked to the accumulation of misfolded α-syn and thereby widely implicated in the pathogenesis of the synucleinopathies [
71,
82]. However, there is still controversy in synucleinopathies regarding the advantageous or detrimental functions of TLRs, especially of TLR4. For example, the lack of TLR4 is associated with αSyn upregulation [
83] and dopamine depletion [
84]. Moreover, Stefanova et al. [
85] showed that the ablation of LR4 prevented αSyn phagocytosis and clearance. However, another study using TLR4 knockout mice revealed less neuroinflammation and neurodegeneration [
86]. Prolonged inflammation can promote αSyn misfolding but many other factors can contribute to the αSyn pathology. Synucleinopathies, including PD, arise from a combination of genetic and non-genetic factors. Processes ranging from neurons and glia interaction to protein-protein interaction and protein misfolding are known to cause neuronal death. Moreover, multimorbidities such as multiple proteinopathies and co-occurring vascular and metabolic dysfunctions together with aging, sex and genetic factors contribute to the neurodegeneration and influence the course of disease [
87]. The presence of combinated proteinopathies and comorbidities has important implications for the research of novel biomarkers and for development of therapeutic targets and strategies [
88].
Although most studies have focused on TLR2 and TLR4, this could also be true for other TLRs, including TLR7 and TLR9 [
89]. In fact, TLR7 activation can lead to the accumulation of αSyn in the brain and TLR9 might also be involved in the activation of microglia and the production of pro-inflammatory cytokines in synucleinopathies [
90]. Additionally, TLR7 inhibition has been shown to reduce the accumulation of αSyn in animal models of PD, suggesting that TLR7 may be a potential therapeutic target for the treatment of synucleinopathies [
91]. In addition, TLR8 targeting by small molecule agents is proposed to have a potential clinical application [
91,
92].
αSyn activates leucine-rich-repeat and pyrin-domain-containing3 (NLRP3) inflammasome, generating extensive microgliosis [
93]. In this scenario, TLRs attend both as “assembling signals” and as an “activation signal” for NLRP3 inflammasome activation responsible for pro-inflammatory cytokine release and thereby microgliosis and astrogliosis. It has been shown that αSyn aggregates released by injured neurons are recognized by TLR2 or TLR4 and can take divergent paths: they can be either moved to lysosome for degradation and clearance or promote the activation of NLRP3 inflammasome causing a diffuse αSyn proteotoxicity in several brain regions such as midbrain, hippocampus and cortex [
94,
95]. In the CNS, the presence of TLRs on the surface of “sentinel cells” such as microglia, neurons, and astrocytes, favors the intracellular uptake, transport, and degradation via lysosomal pathway. However, a partial degradation leads to the further intracellular accumulation and neuroinflammation [
96].
The αSyn proteostasis in the nervous central system is controlled by a selective autophagy pathway termed “synucleinphagy” that also requires the presence of TLRs. TLR-mediated activation of microglia could engulf αSyn into autophagosomes to be degraded; therefore, the disruption of synucleinphagy can cause the accumulation of misfolded αSyn and neuronal death. Various conformations of extracellular αSyn, including monomers, oligomers, and high-molecular-weight aggregates, can induce microglial neuroinflammation via TLRs [
97]. In particular, both TLR2 and TLR4 can interact with αSyn, promoting its internalization into microglia [
85,
98,
99]. Furthermore, targeting TLR2 has proven to be a good approach for inhibiting pathogenic cell-to-cell αSyn transmission and astroglial inflammatory responses, and for clearing toxic species via autophagic machinery. Once internalized, αSyn oligomers spread from neuron to glial cells, engaging and activating TLRs on nearby microglia surface and directly modulating its uptake and intracellular trafficking. Therefore, targeting TLRs and reactive microglia may be a promising therapeutic strategy also for preventing cell-to-cell transmission and slowing the progression of synucleinopathies.
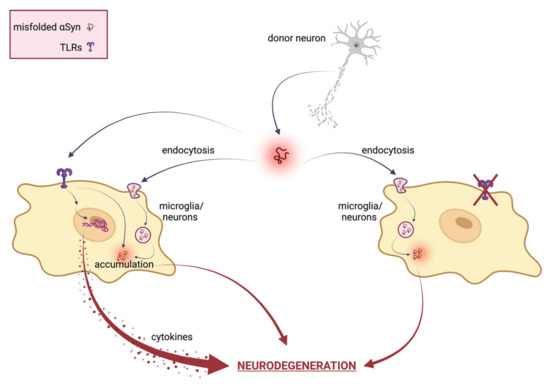
Figure 1 reports a schematic representation of the TLRs’ involvement in αSyn-mediated neurodegeneration.
Figure 1. Schematical representation of TLRs involvement in αSyn mediated neurodegeneration. In neuronal cells, misfolded αSyn can be internalized via TLRs or several indirect endocytosis methods. Extracellular αSyn activates a TLR signaling cascade that results in neurotoxic responses, such as pro-inflammatory cytokine expression and release, ultimately leading to neuronal damage and pathological modification of αSyn. Therefore, TLR specific targeting ameliorates αSyn-mediated neurotoxicity by inhibiting TLR-mediated αSyn internalization and inflammatory response.