Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jason Zhu and Version 1 by Liang Cheng.
Cyclooctyne molecules have found wide applications in the strain-promoted azide–alkyne cycloaddition (SPAAC) reactions, which avoid the biotoxicity caused by the use of Cu(I) catalysts. Among the various cyclooctyne systems, dibenzocyclooctyne (DBCO) series have displayed the highest reaction activity.
- aza-dibenzocyclooctyne
- biarylazacyclooctynone
- synthesis
1. Synthesis Approaches for DIBAC
DIBAC was initially developed by van Delft, [8][1] who used 2-iodobenzyl alcohol 1 and 2-ethynylaniline 2 as the starting materials (Figure 1). A Sonogashira coupling reaction was performed to construct the internal alkyne 3, followed by the protection of the aniline and semi-hydrogenation catalyzed by Pd/BaSO4 to deliver the cis-alkene 5. The terminal hydroxyl methyl was oxidized to aldehyde using the Dess–Martin oxidation reaction and then passed through a reductive amination reaction with the intramolecular amino group to obtain the key intermediate 7. The synthetic route contained multiple steps, however, each step exhibited a relatively high yield, and the total yield for preparing intermediate 7 can reach as high as 70%.
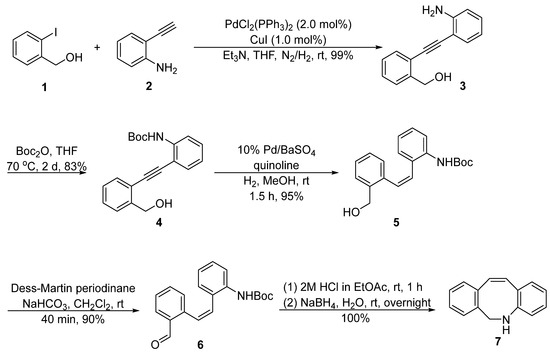
Figure 1. Synthetic approach of intermediate 7 by van Delft.
With the key intermediate 7 in hand, the most commonly used method for synthesizing DIBAC could be achieved in three steps (Figure 2). A simple alkylation would introduce functional probes (azide, alkyne, biotin, etc.) onto the nitrogen under various conditions. Following this, bromination and base-prompted elimination would then give the corresponding product, DIBAC.
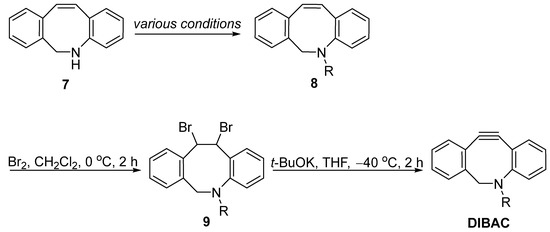
Figure 2. Synthesis of DIBAC from intermediate 7 through bromination and elimination.
Compared with van Delft’s route, Popik et al. developed another synthesis route with fewer steps (Figure 3) [31][2]. Dibenzosuberone 10, a commonly fused tricyclic framework, was used as the starting material. It was converted to oxime 11 and then treated by polyphosphoric acid, which functioned both as an acid catalyst and a dehydrating agent, at 125 °C for 1 h to afford the amide 12 in 97% yield. A selective reduction in the amide linkage with lithium aluminum hydride would then deliver the expected amine 7 in 91% yield. Although there were fewer steps in Popik’s pathway, the overall yield was not superior (40% for three steps from 10).
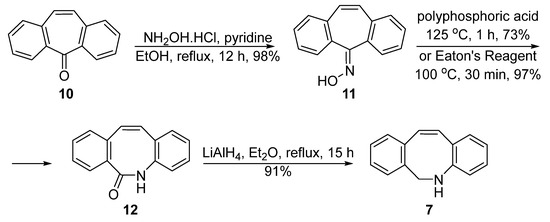
Figure 3. Popik’s improvement of the synthesis of DIBAC.
The key step in the Popik’s route is the Beckmann rearrangement, which was promoted by polyphosphoric acid under a high temperature. Adronov et al. believed that the poor solubility of the polyphosphoric acid may be the reason for the low yield of this step, and therefore used the Eaton reagent instead for this key rearrangement [32][3]. It turned out that the 10 wt% of phosphorus pentoxide solution in methanesulfonic acid [33][4] is an excellent alternative to polyphosphoric acid for the nitrilium ion formation, with which the intermediate 7 could be obtained in 90% yield, even in gram-scale processes (Figure 3).
In 2013, Schubert et al. reported a different method for constructing the DIBAC skeleton using Pd-catalyzed cyclization (Figure 4) [34][5]. This method relied on the regioselectivity of the intramolecular Heck reaction between 2-vinylbenzaldehyde 14 and 2-bromoaniline 13. The former intermediate was prepared via a vinylation of bromo-benzaldehyde by Suzuki coupling with vinylboroxine [35][6] or via the Hiyama reaction with vinylsiloxane [36][7]. A subsequent reductive imine condensation with the 2-chloroaniline 13 afforded the N-benzylaniline 15 containing a vinyl and halide moiety, which then underwent an intramolecular Suzuki–Heck coupling to obtain the key intermediate 7.
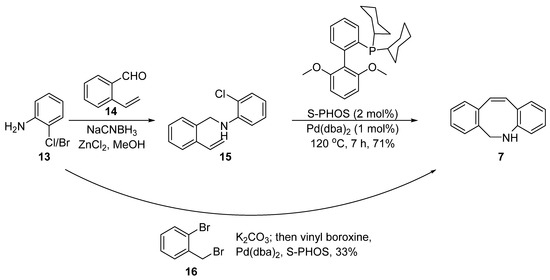
Figure 4. Synthesis of intermediate 7 via Suzuki–Heck coupling.
Considering the modular nature of this method, Schubert further extended it to a one-pot synthesis (Figure 4). They used commercially available 2-bromo-benzyl bromide 16 and 2-chloroaniline 13 as the starting materials. The mixture was subjected to the standard intramolecular amination and Suzuki–Heck coupling and the final product 7 was obtained in 33% yield. The yield was a little lower than that of the two step route due to unavoidable double alkylation of the amino group, and thus further research and optimization may be required.
Unlike the strategies mentioned above that involved dibromination and dehydrohalogenation to generate the triple bond within the dibenzosuberane, Pietzsch et al. utilized ultraviolet irradiation to promote the elimination of carbon monoxide from the 2-cyclopropen-1-one motif, resulting in the release of the C–C triple bond (Figure 5) [37][8]. The key step was the ring closure with tetrachlorocyclopropene 21. Starting from 3,4,5-trimethoxyaniline 17 and 3-methoxybenzaldehyde 18, a reductive amination delivered the N-benzylaniline 19, followed by a dual Friedel–Crafts reaction with tetrachlorocyclopropene 21 to produce the key intermediate 22.
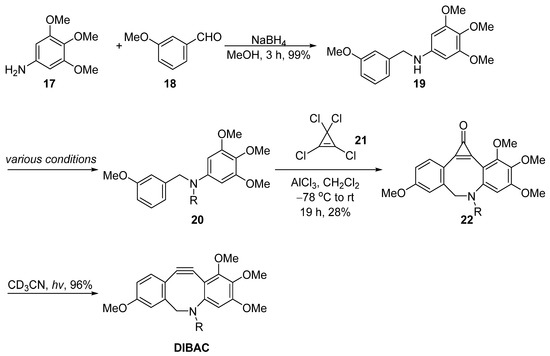
Figure 5. Synthesis of DIBAC by Pietzsch.
While van Delft’s method has been commonly used for the synthesis of DIBAC, recently Hosoya et al. designed new alkyne−cobalt complexes to prepare DIBAC and its derivatives (Figure 6) [38][9]. This strategy is based on the utilization of Pictet−Spengler reaction, in which N-methoxymethyl amide 24 was generated from o-iodoaniline 23 through amidation and N-methoxymethylation. A Sonogashira coupling reaction was performed to construct diarylacetylene 25 that was later treated with Co2(CO)8 to yield an alkyne−cobalt complex 26. The Pictet−Spengler reaction was applied to afford the DIBAC−cobalt complex 27. Finally, the cobalt complex 27 was disassociated using the Me3NO/pyridine/air system to deliver the DIBAC derivative 28. The overall yield was 72% (six steps), which is higher than van Delft’s synthesis method.
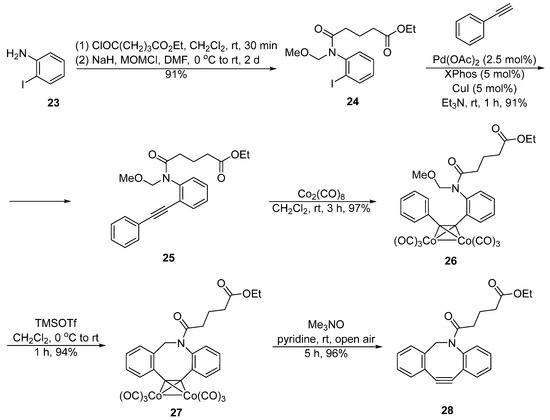
Figure 6. Synthesis of DIBAC by Hosoya.
2. Synthesis Approaches for BARAC
BARAC is another useful cyclooctyne probe, which displays a constant reaction rate higher than that of DIBAC. However, its synthesis methods are quite limited. Bertozzi et al. subjected the Fischer indole intermediate from 1-indanone 29 and phenylhydrazine to an acid-promoted rearrangement to obtain the eight-membered skeleton 30 (Figure 7). The key intermediate was then alkylated with the corresponding linkage and installed in the TMS group as a placeholder. The decorated 31 was subjected to oxidative cleavage with m-CPBA and then converted to the triple bond precursor TMS substituted vinyl trifluoromethanesulfonate 33, which was then cleaved using fluoride ion to generate the BARAC.
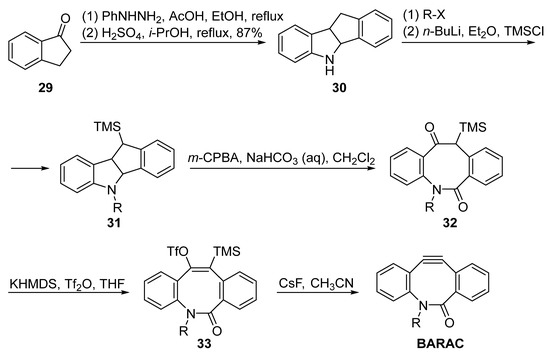
Figure 7. Synthesis of BARAC by Bertozzi.
In 2019, Okano et al. improved Bertozzi’s synthesis method (Figure 9) [39][10]. Instead of introducing a TMS group onto 34, the eight-membered skeleton was firstly subjected to an oxidative cleavage of an amide–ketone 35. Using the KHMDS and PhNTf2 as the dehydrogenation combination, the target BARAC product was obtained in good yields (74% when R = Me). However, when the final step was conducted at 0 °C or higher, only trace amount of the product was observed.
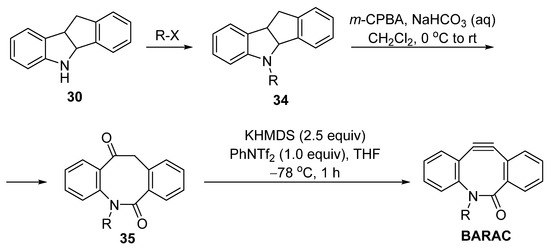
Figure 8.
Okano’s optimization for the synthesis of BARAC.
As mentioned before, an amide intermediate 12 was generated during the synthesis of DIBAC via the Beckmann rearrangement (Figure 3). Therefore, Adronov et al. considered using this process to prepare BARAC derivatives (Figure 9) [32][3]. Similar to the synthesis method of DIBAC, they first prepared the alkylated amide and then subjected the 36 to dibromination to obtain 37, followed by base-facilitated elimination. However, the corresponding BARAC was generated in less than 10% (NMR and TLC) and could only be trapped by click reaction with benzyl azide. Therefore, further optimization was still required.
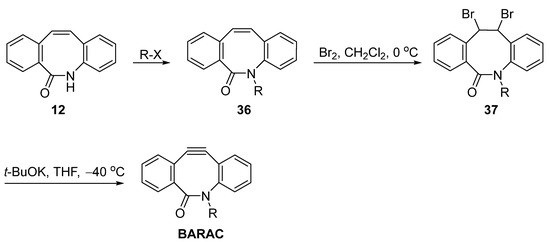
Figure 9.
Adronov’s synthetic route via the amide to BARAC.
References
- Debets, M.F.; van Berkel, S.S.; Schoffelen, S.; Rutjes, F.P.J.T.; van Hest, J.C.M.; van Delft, F.L. Aza-dibenzocyclooctynes for fast and efficient enzyme PEGylation via copper-free (3 + 2) cycloaddition. Chem. Commun. 2010, 46, 97–99.
- Kuzmin, A.; Poloukhtine, A.; Wolfert, M.A.; Popik, V.V. Surface functionalization using catalyst-free azide–alkyne cycloaddition. Bioconjug. Chem. 2010, 21, 2076–2085.
- Adronov, A.; Chadwick, R.; Van Gyzen, S.; Liogier, S. Scalable synthesis of strained cyclooctyne derivatives. Synthesis 2014, 46, 669–677.
- Eaton, P.E.; Carlson, G.R.; Lee, J.T. Phosphorus pentoxide-methanesulfonic acid. Convenient alternative to polyphosphoric acid. J. Org. Chem. 1973, 38, 4071–4073.
- Jäger, M.; Görls, H.; Günther, W.; Schubert, U.S. Pd-Catalyzed Ring Assembly by Vinylation and Intramolecular Heck Coupling: A Versatile Strategy Towards Functionalized Azadibenzocyclooctynes. Chem.-Eur. J. 2013, 19, 2150–2157.
- Slugovc, C.; Perner, B.; Stelzer, F.; Mereiter, K. “Second Generation” Ruthenium Carbene Complexes with a cis-Dichloro Arrangement. Organometallics 2004, 23, 3622–3626.
- Denmark, S.E.; Butler, C.R. Vinylation of aryl bromides using an inexpensive vinylpolysiloxane. Org. Lett. 2006, 8, 63–66.
- Starke, F.; Walther, M.; Pietzsch, H.-J. A Novel Dibenzoazacyclooctyne Precursor in Regioselective Copper-Free Click Chemistry. Arkivoc 2010, 2010, 350–359.
- Sakata, Y.; Nabekura, R.; Hazama, Y.; Hany, M.; Nishiyama, T.; Kii, I.; Hosoya, T. Synthesis of Functionalized Dibenzoazacyclooctynes by a Decomplexation Method for Dibenzo-Fused Cyclooctyne–Cobalt Complexes. Org. Lett. 2023, 25, 1051–1055.
- Hioki, Y.; Itoh, M.; Mori, A.; Okano, K. One-Pot Deprotonative Synthesis of Biarylazacyclooctynones. Synlett 2020, 31, 189–193.
More