PTM refers to irreversible or reversible covalent processing in some proteins after the translation
[13]. It occurs at the amino acid side chains, C-terminus or N-terminus
[14]. PTM changes the properties of amino acids by adding some particular chemical groups, proteins, carbohydrates or lipids to the amino acid side chains, or cleaving bonds enzymatically, which enhances the diversity of protein structures and functions
[13]. These modifications are often induced by enzymatic catalysis, playing a critical regulatory role in physiological and pathological conditions
[15]. Several PTMs are involved in the regulation of NLRP3 inflammasome activation, including ubiquitination, phosphorylation, SUMOylation, alkylation, S-nitrosylation, S-glutathionylation and acetylation
[16].
2. NLRP3 Inflammasome Activation
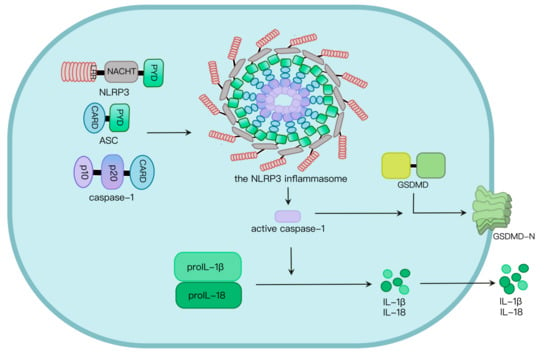
Two signals are required for NLRP3 inflammasome activation. Signal I involves the priming signal, induces IL-1β expression and upregulates NLRP3 expression through activating TLR and NF-κB pathways
[17][18][17,18] as well as NLRP3 phosphorylation. In addition, signal II, transduced by PAMPs and host-derived DAMPs, triggers the assembly and activation of the NLRP3 inflammasome
[19]. Mechanisms by which the NLRP3 inflammasome is activated are extensively explored. At least four models were proposed: K
+ efflux
[20], the generation of mitochondrial reactive oxygen species (mROS)
[21], cathepsin B release from damaged lysosomes
[22] and Ca
2+ mobilization
[23]. NLRP3 oligomerization via its NACHT domain leads to PYD clustering, which elicits recruitment and clustering of ASC through PYD–PYD interaction. ASC clustering subsequently provokes caspase-1 recruitment and assembly of the inflammasome complex. Then, caspase-1 undergoes autocleavage and formation of active p10/p20 tetramer, which cleaves proinflammatory cytokines such as IL-1β and IL-18 into their active molecules
[24]. Active caspase-1 also cleaves GSDMD. The GSDMD N-terminal fragment (GSDMD-N) oligomerizes in the plasma membrane to generate approximately 21 nm-diameter GSDMD pores, leading to osmotic imbalance and cell death called pyroptosis (
Figure 1)
[24][25][24,25].
Figure 1. NLRP3 inflammasome activation. NLRP3, along with ASC, induces caspase-1 activation via assembly of the NLRP3 inflammasome upon challenges. Caspase-1 activation leads to maturation and secretion of proinflammatory cytokines such as IL-1β and IL-18, as well as programmed cell death called pyroptosis induced by GSDMD pores in the plasma membrane.
3. Regulation of Ubiquitination and Deubiquitination in NLRP3 Inflammasome Activation
Ubiquitin, a highly conserved small regulatory eukaryotic protein, contains 76 amino acids and 7 lysine residues, including K6, K11, K27, K29, K33, K48 and K63. It can be covalently attached to target proteins through a cascade of enzymatic reactions catalyzed by ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2) and ubiquitin ligases (E3) [26]. Ubiquitin is bound to the target substrates via an isopeptide bond formed between the C-terminal glycine of ubiquitin and the ε-amino group of lysine in the substrate [27]. Similar isopeptide bonds can be formed from linkage of the C-terminus of one ubiquitin to one of the seven lysine residues or the N terminal methionine on another ubiquitin to form ubiquitin chains [28]. Deubiquitinases remove conjugated ubiquitin from the substrates [29].
3.1. Ubiquitination of NLRP3
NLRP3 ubiquitination plays a critical role in the regulation of its activity. The effects of different types of ubiquitin chains on the NLRP3 activity vary. Among the three enzymes participating in ubiquitination, E3 ubiquitin ligases act as the major proteins involved in the orchestration of NLRP3 inflammasome activity. Its regulation in NLRP3 inflammasome activation is mainly mediated by K48-, K63- and K27-linked ubiquitination. K48-linked ubiquitin chains employ a closed conformation with the hydrophobic residues at the interdomain interface exposed to ubiquitin chain recognition factors, serving as a signal for degradation by the 26S proteasome
[30][51]. K63-linked ubiquitin chains act as a signal for altering the functions of the modified protein, including signaling transduction, DNA repair and intracellular trafficking
[31][52].
3.2. Deubiquitination of NLRP3
Deubiquitinases remove K48- and K11-linked ubiquitin chains, contributing to NLRP3 inflammasome activation. This is consistent with the effect of K48-linked ubiquitination on NLRP3 inflammasome activity. Upon infection with the DNA virus herpes simplex virus type 1 (HSV-1), the stimulator of interferon genes (STING) binds to NLRP3, attenuates K48- and K63-linked ubiquitination of NLRP3 and increases its protein expression on the endoplasmic reticulum, facilitating NLRP3 inflammasome activation and the subsequent release of proinflammatory cytokines
[32][44]. Ubiquitin-specific peptidase 1 (USP1)-associated factor 1 (UAF1, also called WDR48 or p80) is a stoichiometric binding partner of USP1
[33][68]. The UAF1-USP1 deubiquitinase complex selectively eliminates K48-linked ubiquitin chains of NLRP3 to stabilize its expression via interaction with the LRR and NACHT domains, which in turn promotes NLRP3 inflammasome activation
[34][45].
3.3. Ubiquitination and Deubiquitination of ASC and Caspase-1
The influence of K63-linked chains on ASC still remains to be investigated. Both the addition and elimination of K63 ubiquitin chains promote NLRP3 inflammasome activation. This may be associated with different modification sites or regulation by other signaling pathways triggered by ubiquitin enzymes or deubiquitinases. On the one hand, mitochondrial antiviral signaling protein (MAVS) facilitates interaction between the tumor necrosis factor receptor-associated factor (TRAF3) and ASC, provoking K63-linked ubiquitination of ASC at Lys174 and contributing to NLRP3 inflammasome activation
[35][36][48,72]. Pellino E3 ubiquitin protein ligase 1 (Peli1) mediates both K48- and K63-ubiquitination
[37][38][73,74].
4. Regulation of Phosphorylation and Dephosphorylation in NLRP3 Inflammasome Activation
Phosphorylation modulates protein function and controls the turnover of its targets and subcellular localization by altering protein conformation or influencing protein–protein interaction. Protein kinases mediate the phosphate group transfer from ATP to serine, threonine and tyrosine residues of the substrates, while phosphatases removes the phosphate group of a phosphorylated protein substrate
[39][80]. Phosphorylation and dephosphorylation of the components of the NLRP3 inflammasome control its activity.
4.1. NLRP3 Phosphorylation
NLRP3 phosphorylation at Ser725
[40][81], Ser194
[41][82] or tyrosines in the PYD-NACHT polybasic linker
[42][89] contribute to NLRP3 inflammasome activation. Misshapen-like kinase 1 (MINK1), a member of the mammalian germinal center kinase (GCK) family
[43][90], binds to the NLRP3 LRR domain, and phosphorylates NLRP3 at Ser725, which are critical for the priming step of NLRP3 inflammasome activation
[40][81]. ROS serves only as a priming signal, but fails to contribute to the activation step of the NLRP3 inflammasome
[44][91]. It is able to increase the kinase activity of MINK1 and facilitate NLRP3 phosphorylation at Ser725. This subsequently promotes inflammasome priming
[40][81].
4.2. NLRP3 Dephosphorylation
NLPR3 dephosphorylation at Ser5
[45][84], Ser803
[46][95] or Tyr861
[47][85] enhances NLRP3 inflammasome activation. The phosphatase protein, phosphatase 2A (PP2A), dephosphorylates NLRP3 at Ser5 to allow its activation, and the regulation of PP2A in NLRP3 inflammasome activation is controlled by BTK
[45][48][84,96]. Protein tyrosine phosphatase non-receptor type 22 (PTPN22) interacts with and dephosphorylates NLRP3 at Tyr861, decreasing NLRP3 inflammasome-mediated IL-1β secretion
[47][85].
4.3. Phosphorylation of ASC and Caspase-1
The phosphorylation and dephosphorylation of NLRP3 at different sites play distinct roles in the regulation of NLRP3 inflammasome activation, while the phosphorylation of ASC and caspase-1 promotes NLRP3 inflammasome activation. Pyruvate kinase (Pyk2)
[49][87] and JNK
[50][99] are involved in human ASC Tyr146 (mouse ASC Tyr144) phosphorylation, facilitating ASC oligomerization and NLRP3 inflammasome assembly. Spleen tyrosine kinase (Syk) contributes to NLRP3 agonist-mediated ASC self-association and inflammasome assembly through phosphorylating ASC at Tyr146 and Tyr187 residues
[50][51][99,100]. LPS treatment induces interaction between p21 (Rac family small GTPase 1) activated kinase 1 (PAK1) and caspase-1, mediating caspase-1 phosphorylation at Ser376 and its activation
[52][88].
5. Regulation of SUMOylation in NLRP3 Inflammasome Activation
The small ubiquitin-like modifier (SUMO) protein is evolutionarily conserved and ubiquitously expressed in eukaryotes. It belongs to the ubiquitin-like family and alters the properties and functions of modified proteins via PTMs
[53][54][55][101,102,103]. Four SUMO proteins have been identified in humans, SUMO1–4. SUMO2 and -3 are highly homologous
[56][104]. SUMOylation is a reversible PTM process. SUMO binds to a lysine residue of a substrate and is removed from the modified protein through SUMO-specific peptidase-mediated deSUMOylation. SUMO is expressed as a C-terminally extended precursor, and then processed to generate the active form. The SUMO-activating enzyme SAE1/SAE2 covalently links to the C-terminus of SUMO via the sulfhydryl group of a cysteine residue; then, SUMO is transferred to the SUMO-conjugating enzyme Ubc9, and finally conjugated to a lysine side chain of the target protein mediated by a SUMO ligase. SUMO chains are able to assemble on substrates
[57][58][105,106].
NLRP3 Lys204 SUMOylation facilitates its activation
[59][107], while Lys689 SUMOylation suppresses its activation
[60][108]. Ubc9 binds to NLRP3 to mediate its SUMOylation at Lys204 induced by SUMO1, which facilitates ASC oligomerization and inflammasome assembly. Interaction between SUMO-specific protease 3 (SENP3) and NLRP3 results in NLRP3 deSUMOylation and attenuates ASC speck formation, as well as inflammasome activation
[59][107]. Mitochondrial-anchored protein ligase (MAPL), also called mitochondrial E3 ubiquitin protein ligase 1 (MUL1), mediates RING-finger-dependent NLRP3 Lys689 SUMOylation, and enhances caspase-1 maturation and IL-1β release in murine macrophages and human cells. NLRP3 Lys689 deSUMOylation induced by SENP6 and 7 promotes NLRP3-dependent ASC oligomerization and caspase-1 activation. MAPL has no effect on absent in melanoma 2 (AIM2) inflammasome activation
[60][108].
6. Regulation of Alkylation in NLRP3 Inflammasome Activation
Alkylation targeting ATPase activity of NLRP3 negatively controls the inflammasome activation through a decreasing affinity for ATP. The NACHT domain, carrying ATPase activity, adopts an ADP/ATP switch mechanism to orchestrate NLRP3 activation
[61][110]. The NACHT domain is composed of four subdomains, including a nucleotide-binding domain (NBD, aa131–373 in human NLRP3), a helical domain 1 (HD1, aa374–434), a winged-helix domain (WHD, aa435–538) and helical domain 2 (HD2, aa539–676). Interaction between WHD His522 and ADP keeps NLRP3 in a closed and inactive conformation
[62][63][111,112]. Treatment with NLRP3 agonists leads to the exit of ADP, the binding of ATP with the Walker A motif in NBD and ATP hydrolysis
[61][110]. MCC950 suppresses ATP hydrolysis and NLRP3 inflammasome activation via interaction with the Walker B motif in NBD
[64][113]. 3, 4-methylenedioxy-β-nitrostyrene (MNS)
[65][114], 2-cyclohexylimino-6-methyl-6,7-dihydro-5H-benzo
[1][3][1,3], oxathiol-4-one (BOT-4-one)
[66][115] and vanillylacetone
[67][116] mediate NLRP3 alkylation and reduce its ATPase activity.
7. Regulation of S-Nitrosylation in NLRP3 Inflammasome Activation
Similar to NLRP3 alkylation, NLRP3 S-nitrosylation plays an inhibitory role in the inflammasome activity. S-nitrosylation refers to the covalent binding of a NO group to a protein cysteine thiol to form S-nitrosothiols
[68][125]. S-nitroso-N-acetylpenicillamine (SNAP), an NO donor, dampens nigericin-induced capase-1 maturation, as well as release of IL-1β and IL-18, in the TLR1/2 agonist PAM3CSK4-primed murine peritoneal macrophages. Priming with PAM3CSK4 causes S-nitrosylation of NLRP3 and caspase-1, and the C-terminus of NLRP3 is more susceptible to S-nitrosylation than its N-terminus. Inflammasomes of AIM2 and NLRC4 are partially inhibited
[69][126]. HEK293T cells transfected with NLRP3 or caspase-1 expression plasmids were treated with SNAP or not, and lysed to obtain lysate 1. Independent cultures transfected with plasmids expressing the other components of the NLRP3 inflammasome and IL-1β were lysed to obtain lysate 2. Mature IL-1β was assessed in the mixed lysates with lysate 1 and lysate 2.
8. Regulation of Acetylation in NLRP3 Inflammasome Activation
In contrast to alkylation and S-nitrosylation of NLRP3, NLRP3 acetylation boosts the inflammasome activation. Lysine acetyltransferase 5 (KAT5), also called Tat-interactive protein 60 kDa (TIP60), belongs to MOZ-Ybf2/Sas3-Sas2-TIP60 (MYST) family
[70][129]. KAT5 mediates NLRP3 acetylation at Lys24, facilitating its interaction with NEK7 and oligomerization. The KAT5 inhibitor NU9056 restricts NLRP3-dependent caspase-1 maturation and IL-1β release both in vivo and in vitro
[71][130]. Sirtuin 2 (SIRT2), an NAD
+-dependent deacetylase and a metabolic sensor, targets NLRP3 for deacetylation in BMDMs.
Sirt2 deletion or treatment with the SIRT2 inhibitor AGK2 results in increased IL-1β production and cleaved caspase-1, but has no effect on pro-IL-1β expression.
Sirt2 knockout makes no change to caspase-1 maturation and IL-1β secretion following stimulation with flagellin, a NLRC4 inducer, or poly(dA:dT), an AIM2 inducer.
9. Regulation of S-Glutathionylation in NLRP3 Inflammasome Activation
Protein S-glutathionylation, an oxidative PTM, is a reversible formation of mixed disulfides between tripeptide glutathione and low-pKa cysteine
[72][132]. Glutathione transferase Omega 1 (GSTO1-1) belongs to the cytosolic glutathione transferase (GST) super family
[73][133].
Gsto1-1 deletion reduces proinflammatory cytokine expression and ameliorates the inflammatory response in response to LPS in mice. GSTO1-1 deglutathionylates NEK7 Cys253 to promote its interaction with NLRP3 and the inflammasome activation
[74][134]. Superoxide dismutase 1 (SOD1) contributes to caspase-1 activation via inhibiting its glutathionylation. Upon stimulation with LPS + ATP, SOD1 deficiency leads to increased ROS generation, decreased cellular redox potential and reversible oxidation and glutathionylation of caspase-1 Cys397 and Cys362, eliciting the inhibition of caspase-1 activity. Caspase-1 is activated and not glutathionylated in WT murine peritoneal macrophages
[75][135].
10. The NLRP3 Inflammasome and Cancers
The NLRP3 inflammasome plays dual roles in the pathogenesis of cancers. It has a protective anti-tumorigenic effect in colitis-associated cancer, colorectal cancer, hepatocellular carcinoma and melanoma, and plays a pro-tumorigenic role in breast cancer, colon cancer, colorectal cancer, epithelial skin cancer, fibrosarcoma and gastric cancer
[76][77][138,139]. Regulation of NLRP3 inflammasome activity via PTMs is essential for cancer development and progression. TRIM31 is upregulated at the protein level in human hepatocellular carcinoma and colorectal cancer, and promotes invasion and metastasis
[78][79][140,141]. It mediates the K48-linked ubiquitination and degradation of NLRP3, restricting NLRP3 inflammasome activity
[80][35]. Alpinumisoflavone and estrogen suppress the proliferation and metastasis of hepatocellular carcinoma cells via enhancing NLRP3 inflammasome activation
[81][82][142,143]. Caffeic acid phenethyl ester (CAPE) enhances NLRP3 ubiquitination via facilitating NLRP3–Cullin1 interaction and suppresses NLRP3 inflammasome activation, which protects mice from azoxymethane/dextran sulfate sodium-induced colon cancer
[83][84][39,144].