Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Jinyi Tan and Version 2 by Jessie Wu.
Magnaporthe oryzae (synonym of Pyricularia oryzae), which causes rice blast disease, is a plant pathogenic fungus belonging to the Ascomycota phylum.
- Magnaporthe oryzae
- plant fungal pathogen
- rice blast
1. Introduction
In 1879, Magnaporthe oryzae was initially named Trichothecium griseum by Cooke without a detailed description [1]. In the following century, several names were given to the fungus based on the teleomorph and anamorph stages, such as Ceratosphaeria grisea by Herber (1971) and Dactylaria oryzae by Sawada (1917). In 1990, based on host specificity, physiological differences, and genetic evidence, Rossman corrected its name to Pyricularia oryzae [2]. P. oryzae was used to refer to the asexual stage, and the sexual stage was named Magnaporthe grisea by Couch and Kohn (2002). Additional phylogenetic analysis and interstrain fertility tests showed that M. grisea should be assigned to Digitaria (crabgrass)-infecting isolates, whereas M. oryzae causes damage to rice and other important crops in the Poaceae family, such as millet and wheat [3].
Magnaporthe oryzae is one of the most devastating agricultural pathogens since it primarily attacks cultivated rice (Oryza sativa), an important staple food feeding over 50% of the world’s population [4][5][4,5]. Its infection can be destructive under favorable conditions, contributing to 10–30% of the annual rice yield loss [6][7][6,7]. Given that it can affect both temperate and tropical rice growth, the disease has been widely distributed in 85 countries with various environmental conditions [7][8][9][7,8,9]. As a consequence, it is ranked the most damaging fungal pathogen in the world [9].
Magnaporthe oryzae can cause disease on rice plants at all developmental stages and can infect different tissues, including leaves, stems, nodes, and panicles [10]. Its aerial conidiophores produce three-celled and teardrop-like conidia that are arranged in a sympodial manner. The attachment of these conidia to the host surface initiates the infection cycle [11]. Conidia adhere to the host with the help of mucilage, a thick and gluey substance stored at the spore tip. They then germinate and generate germ tubes before appressoria differentiation [10][11][12][10,11,12]. The death of three-celled conidia triggers the formation of single-celled appressoria [13]. Then, appressoria become melanized and accumulate glycerol, generating turgor to create pressure for penetration into the host epidermal cells [13]. Upon penetration, it colonizes plants using invasive hyphae that grow intracellularly [14]. Notably, membrane caps, formed on the invading hyphal tips, are used for cell-to-cell movement via manipulating the plasmodesmata [14]. Thus, the membrane interactions between M. oryzae and plant hosts are important for the invasion of hyphae into plant cells [15].
Upon penetration, fungal pathogens can secret effectors to promote virulence and suppress host immunity [16]. In turn, plants have evolved resistance genes (R) that encode R proteins to recognize the presence of effectors and enable effector-triggered immunity (ETI) [16][17][16,17]. In M. oryzae, when the first hyphal cell invades, effectors accumulate in the biotrophic interfacial complex (BIC), the site for effector delivery [15][18][15,18]. Two types of effectors were defined in M. oryzae based on their localization in plant cells: cytoplasmic effectors entering the host cell cytoplasm via the BIC, and BIC-independent apoplastic effectors being delivered into the apoplast [19][20][19,20].
Due to its notable appressoria formation, the secretion of effectors during the invasion, and the effector delivery to the apoplast and cytoplasm of plant cells, M. oryzae has been used as a model plant fungal pathogen to understand pathogen genomics and pathogen–host interactions [11].
2. Genome Sequencing
The first analysis of the M. oryzae genome was published (as M. grisea) in 2005 using the whole-genome shotgun (WGS) approach, with 7× coverage. The genome of M. oryzae 70-15, originally derived from a cross between isolates of rice and weeping lovegrass, consisted of seven chromosomes with an estimated size of 38.8 Mb (GenBank accession number: AACU00000000.3) [21][22][21,22]. However, this draft genome sequence was compromised because of significant retrotransposon content. After improvements in verifying unique sequence anchors, extending contigs and scaffolds, and filling the remaining gaps, the genome sequence of the M. oryzae strain 70-15 was refined at the Broad Institute in 2015 with an estimated size of 41 Mb. The high-confidence annotation and gene models were also generated using multiple methods including RNA-seq data and expressed sequence tags (ESTs) alignments, and homologous gene and Basic Local Alignment Search Tool (BLAST) searches [23]. Therefore, this 41 Mb genome sequence of strain 70-15 became the standard reference for M. oryzae, consisting of seven chromosomes with about 51.6% GC content (BioProject Accession number: PRJNA13840).
The initial analysis of the M. oryzae strain 70-15 sequenced in 2005 revealed a family of G-protein-coupled receptors that are specific to M. oryzae and have an expression in infection-related development [22]. Furthermore, they also unveiled three mitogen-activated protein kinase (MAPK) cascades including appressoria development and penetration peg formation, which are crucial in the plant infection process, as well as the relevant genes involved in these signaling cascades. In addition, a significant number of predicted secretion proteins and putative effector proteins, as well as synthases/synthetases involved in secondary metabolic pathways, were identified [22]. Many of these became targets for reverse genetic analysis.
Subsequently, the genome sequences of many other M. oryzae strains became available. For example, Xue et al. (2012) [24] sequenced two M. oryzae field isolates, Y34 (isolates from China) and P131 (from Japan), using Sanger and 454 sequencing. Genome comparisons revealed that the genome size and overall structure were similar among different strains, but more genes that might be involved in host–pathogen interactions were found in the field isolates [24]. They also found different distributions of transposon-like elements among these three strains, indicating genetic variations [24]. More recently, Dr. T. R. Sharma’s group sequenced the whole genome of two isolates from India, an avirulent isolate RML-29 and a highly virulent isolate RP-2421, and identified several isolate-specific genes and potential effectors through pan-genome analysis, which might be involved in fungal pathogenicity and fungal–plant interactions [25][26][25,26].
Repetitive transposable elements (TEs) may play important roles in both genome and pathogenic variations in M. oryzae [22][24][27][22,24,27]. They may mediate fungal virulence via the inactivation or deletion of pathogen-associated molecular patterns (PAMPs)-encoding genes and can trigger plant defense responses [28][29][28,29]. The M. oryzae genome contains approximately 10% of the repetitive sequences in various isolates [22][23][24][27][22,23,24,27]. Xue et al. (2012) [24] also uncovered that 23.8% of the TE-disrupted genes were predicted to encode signal peptide sequences, highlighting that this percentage was higher than the average percentage of the whole genome. The single-molecule real-time sequencing also revealed that TEs are involved in chromosomal translocation and secreted proteins (SPs) polymorphisms [27]. In addition, transposon and effector-rich mini-chromosomes were observed in the M. oryzae MoT isolate, which contribute to the field adaptation [30]. These findings suggested a significant role of TEs in defining host specificity and fungal virulence.
3. Transcriptomic and Secretome Analysis
The availability of whole-genome sequencing data has facilitated the transcriptomic and secretome analysis of M. oryzae. The first comprehensive genome-wide transcriptional profiling study of M. oryzae was carried out using microarray analyses. Changes in genome-wide gene expression during the early stages of spore germination and appressoria formation were identified [31]. A microarray analysis of the fungal-invading hyphae at an early stage (36 h post inoculation) uncovered several putative effectors, including fungal biotrophy-associated secreted (BAS) proteins [20]. Moreover, microarray data of invasive growth on rice and barley at a later stage (72 hpi) also revealed multiple genes associated with stress responses and invasive growth [32].
In recent years, high-throughput RNA sequencing has become an efficient approach to provide high-quality transcriptome data. Many transcriptomic analyses were carried out to study host–pathogen interactions. For example, Kawahara et al. (2012) [33] investigated the mixed transcriptome of the pathogen M. oryzae and host O. sativa, revealing 240 upregulated genes encoding potential secreted proteins and many known infection-related genes in M. oryzae [33]. Another study on plant–fungal interactions between the host rice plants and M. oryzae discovered several novel effectors and virulence-related genes, including 98-06 isolate-unique genes IUG6 and IUG9, which were involved in the fungal pathogenicity and located in the BIC [34].
Secretome analysis is another useful approach to identifying crucial genes through studying secreted proteins and their secretion pathways. One M. oryzae secretome study during the early stages of infection identified 53 secreted proteins, including proteins that functioned by modifying fungal lipid and cell walls, detoxifying reactive oxygen species, as well as encouraging fungal metabolism [35]. Liu et al. (2021) [36] also identified several secreted proteins requiring N-glycosylation, which play essential roles in fungal pathogenicity and cell wall integrity [36]. Combining transcriptomic and secretome analyses, these datasets provide an abundant reservoir of candidates for reverse genetic analysis to help understand the molecular mechanisms in M. oryzae–rice interactions.
Mutant analysis is a definitive process used to establish a causal relationship between a gene and a specific biological process. Many methods were applied to generate mutants in M. oryzae, such as T-DNA insertion, RNA interference (RNAi), and homologous recombination (deletion). Among them, gene deletion via homologous recombination has been the most commonly used method in the past few decades. The latest CRISPR technology has also been developed to improve gene-editing efficiency [37][38][37,38]. The rest of this resviearch w will give an overview of all M. oryzae genes studied to date using such approaches. A Venn diagram of the listed genes is provided for readers’ overview (Figure 1). Over 400 genes have been summarized in this resviearchw, and it is clear to see that most of them contribute to both the development and virulence of M. oryzae. A chromosomal map including the significant genes involved in both processes was generated to detect possible cluster patterns and genomic features (Figure 2).
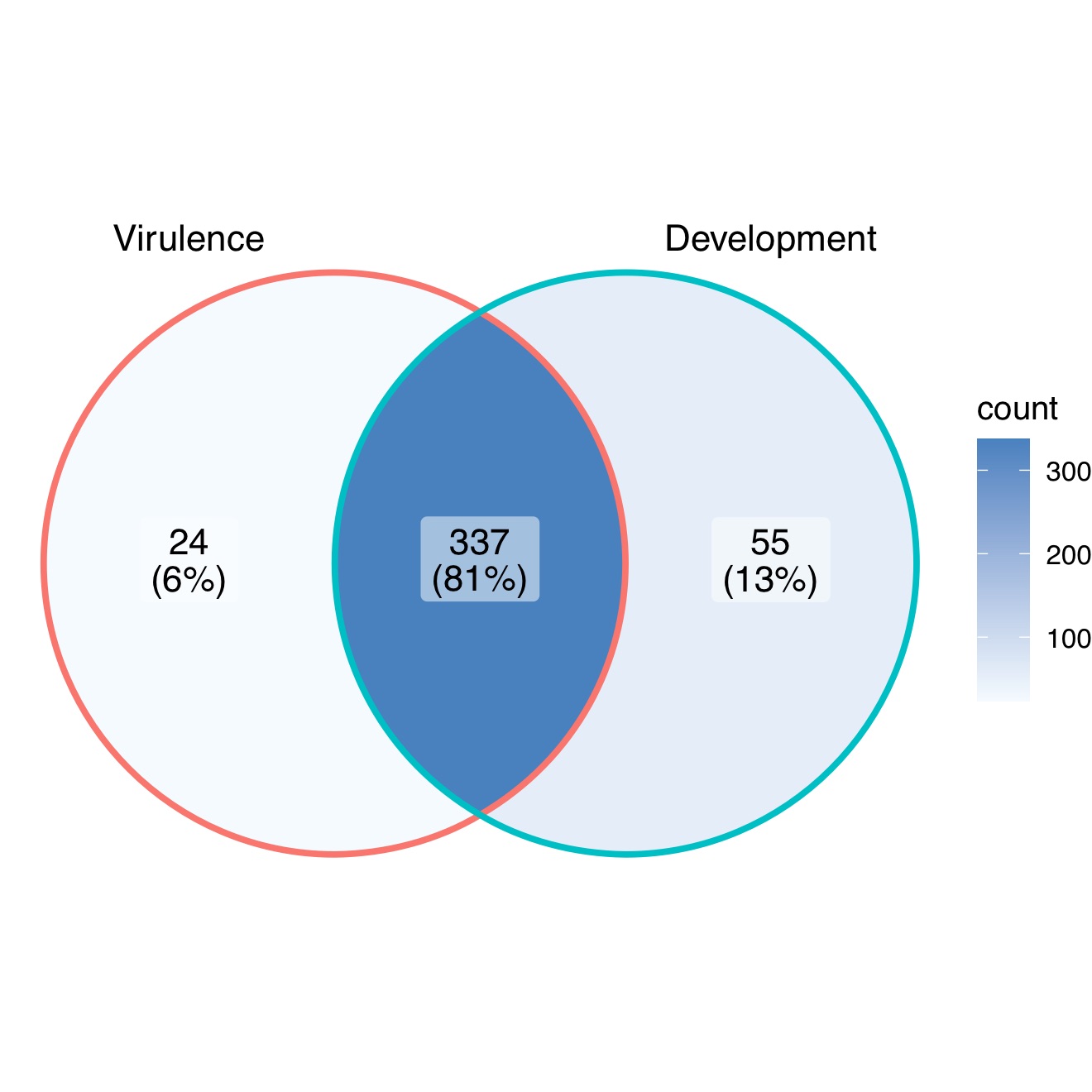
Figure 1.
A Venn diagram summary of
M. oryzae
genes studied with mutant analysis.
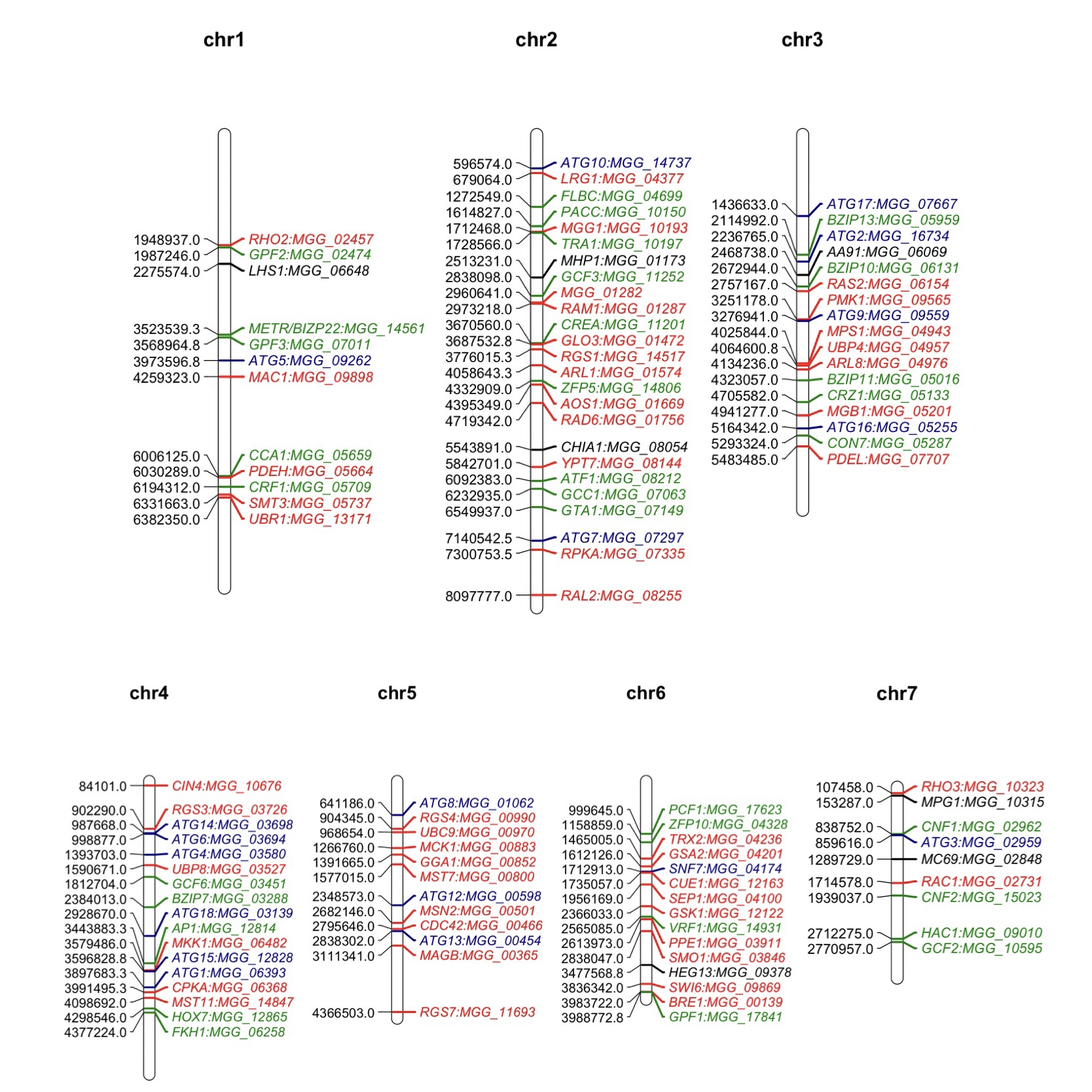
Figure 2. Map positions of important genes studied in the M. oryzae genome through mutant analysis. The numbers on the left of each chromosome represent the locations of these genes. Genes in this map contribute to both development and virulence of M. oryzae. Genes labeled in red are components in different important signaling pathways. Genes labeled in green are transcription factor-encoding genes. Genes labeled in blue are autophagy related while the ones in black are genes encoding effectors. The chromosomal map was drawn using ‘MapChart’ software.