Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ihtisham Ul Haq and Version 2 by Dean Liu.
The genetic variants of HLAs (human leukocyte antigens) play a crucial role in the virus–host interaction and pathology of COVID-19. The genetic variants of HLAs not only influence T cell immune responses but also B cell immune responses by presenting a variety of peptide fragments of invading pathogens. Peptide cocktail vaccines produced by using various conserved HLA-A2 epitopes provoke substantial specific CD8+ T cell responses in experimental animals. The HLA profiles vary among individuals and trigger different T cell-mediated immune responses in COVID-19 infections.
- COVID-19
- pandemic
- genetic susceptibility
- ACE-2
- HLA
1. Introduction
In December 2019, numerous patients in Wuhan, China presented with pneumonia and the causative agent was found to be a novel coronavirus [1][2][3][1,2,3]. Later investigations based on real-time PCR (polymerase chain reaction) led to the identification of a novel coronavirus [4][5][4,5]. Officially, the new coronavirus was discovered on the 7th of January 2020 and given the name nCoV-19 (novel coronavirus 2019) by the WHO (World Health Organization) [6]. The viral nomenclature, ICTV (International Committee on Taxonomy of Viruses) later replaced the nCoV-19 with severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) as it was genetically parallel to the previous SARS coronavirus. The disease caused by SARS-CoV-2 was named COVID-19 (coronavirus disease 2019) officially by the WHO [7]. Owing to the high virulence, pathogenicity, and contagiousness of SARS-CoV-2, in the second week of March 2020, the WHO declared COVID-19 as a pandemic [8]. The COVID-19 outbreak caused by SARS-CoV-2 received a great deal of interest across the world [9]. As of February 21st, 2023, more than 757, 264, 511 cases had occurred from the COVID-19 disease [10].
Since the outbreak of COVID-19, several approaches were proposed for finding potential therapeutics [11][12][13][14][15][16][11,12,13,14,15,16], until the development of effective anti-COVID-19 vaccines [17].
However, crucial questions about individual genetic variability in immune responses, disease susceptibility, and severity, as well as the clinical picture of the pandemic, remain unanswered [18]. In the past, the genetic-based approach of the HLA system, including the HLA-class I and II systems, has been used to trace the etiological agent of newly emerging infectious diseases and its interface with the variations in clinical outcomes of SARS-CoV disease [19]. Given its role in SARS-CoV and other viral diseases, such as MERS CoV (HLA-DRB1*11:01 and DQB1*02:02) [20], influenza (HLA-DRB1*0401 and HLA-DRB1*0402) [21], dengue (HLA-DRB1 * 0901 and HLA-A*24) [18], and hepatitis B (HLA- DPB1*09:01, DPB1*04:01, and DQB1*06:01) [22], the HLA system could also be beneficial in treating COVID-19 infections. The HLA genetic variant encodes hundreds of protein-coding genes that regulate the fundamental molecular and cellular processes, specifically the immune responses [23].
One study found that certain HLA alleles, such as HLA-B46:01 and HLA-B15:03, were associated with a lower risk of severe COVID-19, while other alleles, such as HLA-B07:02, were associated with an increased risk [24]. Another study found that HLA-A02:01, HLA-B07:02, and HLA-C07:29 were associated with a decreased risk of severe COVID-19, while HLA-B35:01 and HLA-C04:01 were associated with an increased risk [25]. These findings suggest that HLA variants may influence the immune response to SARS-CoV-2 infection and contribute to the variability in disease severity observed among individuals. It is currently unclear whether HLA variants arise in response to specific viral stimuli or occur in the context of most viral infections. However, some studies have shown that certain HLA alleles are associated with the protection or susceptibility to other viral infections. For example, HLA-B57:01 has been associated with slower HIV disease progression, while HLA-B15:05 has been associated with susceptibility to hepatitis B virus infection [26]. These findings suggest that HLA variants may be shaped by viral selection pressures.
Furthermore, the ACE-2 receptor has played a significant role in the immune and inflammatory factors associated with COVID-19 pathogenesis. ACE-2, a homolog of ACE, is produced by several human organs and tissues and has a broad spectrum of biological functions [27]. A spike glycoprotein on the coronavirus’s viral envelope adheres to ACE-2 on the membrane of host cells that express ACE-2. After binding, the virus and host cell fuse their membranes, which activates infection by releasing viral RNA into the cytoplasm. Studies have shown that COVID-19 infections can reduce ACE-2 expression on cells, which might cause significant organ damage by disrupting the normal equilibrium between ACE/ACE-2 and Ang-II/angiotensin [28]. This makes ACE-2 a potential target for the development of specific treatments, antibodies, and vaccines related to COVID-19 infection.
2. Angiotensin-Converting Enzyme 2 (ACE-2) in COVID-19
The susceptibility of the host to SARS-CoV-2 infection is determined by the expression of the cellular receptor ACE-2, which varies in different human tissues [29][41]. ACE-2 is highly expressed in adipose tissues, the heart, small intestine, thyroid, and testis, while it is moderately expressed in the lung, adrenal gland, bladder, colon, and liver. This partly explains the cardiac injuries observed in SARS-CoV-2 infected patients [30][42]. On the other hand, lower levels of ACE-2 expression have been reported in the spleen, bone marrow, and white blood cells [30][42]. The detection of SARS-CoV-2 in the stool of infected patients suggests that ACE-2 is expressed in the gastrointestinal tract [31][43]. The broad cellular tropism observed in SARS-CoV-2 infection is due to the wide range of predominant symptoms [30][42]. Although the expression levels of ACE-2 remain consistent in every individual, no significant gender, age, or race-based difference in expression has been reported so far [30][42]. There is a difference between the protein expression of ACE-2 and its mRNA expression level, indicating that ACE-2 is regulated post-transcriptionally [30][42] (Figure 1).
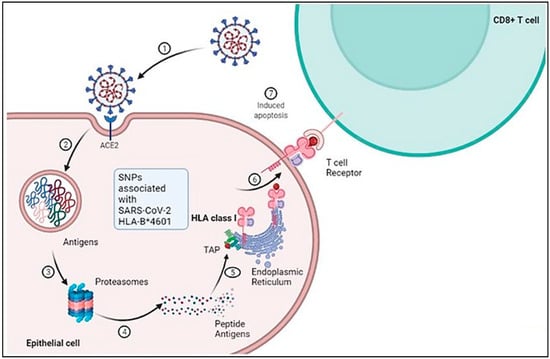
Figure 1. Schematic representation of cellular and molecular pathophysiology of SARS- CoV-2 (COVID-19).SARS-CoV-2 interacts with the ACE-2 receptor resulting in membrane fusion and cytoplasmic entry. 2 and 3: Proteasomal degradation of SARS-CoV-2 in the endolysosome results in antigen generation. 4 and 5: Further processing of the peptide antigens through interaction with HLA class 1 and complexing with the endoplasmic reticulum leads to its presentation on the cell surface. 6 and 7: The processed antigen is further represented by the APC through its β2M receptor, which activates T cells when they encounter the T cell receptors, leading to induced apoptosis. Adapted from “Acute Immune Responses to Coronaviruses”, by BioRender.com (2022). https://app.biorender.com/biorender-templates (accessed on 6 December 2022).
3. Genetic Polymorphism of ACE-2
Recently, genetic polymorphisms in ACE-2 have been reported in various cases, which can affect host susceptibility patterns by altering virus–host interactions. Various variants of ACE-2, such as S19P, T92I, I21V, K26R, E23K, T27A, Q102P, N64K, and H378R, have been found to increase the host’s susceptibility to COVID-19 [32][44]. In addition, ACE-2 expression correlates with different immune signatures, which in turn vary across different populations based on gender, age, and race. These factors can contribute to the high mortality rate associated with COVID-19 [33][45].
SARS-CoV-2 penetrates host cells via angiotensin-converting enzyme 2 (ACE-2), which is abundantly expressed in the heart, kidneys, and lungs and sheds into the plasma. ACE-2 regulates the renin-angiotensin-aldosterone system (RAAS). SARS-CoV-2 disrupts the ACE/ACE-2 balance and activates the RAAS, ultimately, leading to COVID-19 development, particularly in individuals with comorbidities [34][35][46,47]. The SARS-CoV-2 spike protein binds to ACE-2 on the cell surface, leading to the internalization and degradation of ACE-2 and a reduction in its expression. This shift in the balance between ACE and ACE-2, in favor of ACE, can result in increased angiotensin II production and activation of the RAAS, leading to the release of proinflammatory cytokines and contributing to the development of cardiovascular and renal complications in COVID-19.
Angiotensin-converting enzyme 2 (ACE-2) is critical for SARS-CoV-2 infection of a host species, as the S1 protein/receptor interaction is essential for obtaining access to host cells. S1 comprises a receptor-binding domain (RBD), which directly attaches to the peptidase domain (PD) in ACE-2 [36][48]. This is followed by cleavage of the S1 protein, which is achieved through acid-dependent proteolytic cleavage by one or several host proteases, including cathepsins, transmembrane serine protease (TMPRSS2), TMPRSS4, or human airway trypsin-like protease [37][49]. This results in the fusion of viral and cellular membranes. Following fusion with the host membrane, two heptad repeats in the S2 protein form a funnel-like shape in an antiparallel six-helix bundle, allowing fusion and release of the viral genome into the cytoplasm [38][50] (Figure 2). After replication and subgenomic RNA production, the viral structural proteins are translated and inserted into the endoplasmic reticulum (ER), and then, moved through the secretory route to the endoplasmic reticulum–Golgi intermediate, releasing multiple virions [39][51].
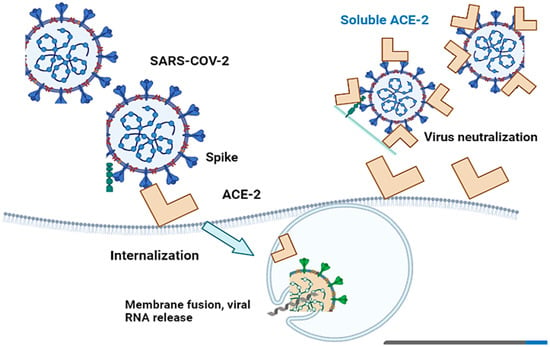
Figure 2. Cell entry of SARS-CoV-2 with ACE-2-mediation and virus infection inhibition by recombinant soluble ACE-2 protein. SARS-CoV-2 penetrates host cells via angiotensin-converting enzyme 2 (ACE-2), which subsequently results in the fusion of viral and cellular membranes.
The crystal structures of the most powerful antibodies (P2C-1F11, P2B-2F6, and P2C-1A3) confirmed competition with ACE-2 binding, demonstrating that inhibiting the RBD and ACE-2 can mediate viral neutralization [40][52]. After screening a wide panel of human mAbs that target the spike protein, two antibodies, COV2-2196 and COV2-2130, were found to identify non-overlapping epitopes on the RBD and bound concurrently to the S protein, synergistically triggering viral neutralization [41][53].