Being initially described as a factor of virally induced leukemias, Fli1 (Friend leukemia integration 1) lately has attracted considerable interest due to its role in both healthy physiology and a variety of pathological conditions. Over the past years Fli1 was found to be one of the crucial regulators of normal hematopoiesis, vasculogenesis and immune responses. However, abnormal expression of Fli1 due to genetic predisposition, epigenetic reprogramming (modifications) or environmental factors is associated with a few diseases of different etiology. Fli1 hyperexpression leads to malignant transformation of cells and progression of cancers such as Ewing’s sarcoma. The deficiency of Fli1 implicates in development of systemic sclerosis and hypertensive disorders, which are often accompanied by pronounced fibrosis in different organs. This work summarizes initial findings and most recent advances in defining the role of Fli1 in diseases of different origin with emphasis on its pro-fibrotic potential.
- Fli1
- fibrosis
- angiogenesis
- systemic sclerosis
- uremic cardiomyopathy
1. Introduction
2. Diseases Associated with Fli1 Deficiency
2.1. Fli1 in Systemic Sclerosis
Fli1 as inductor of fibrosis. Systemic sclerosis (SSc or scleroderma) is a complex and highly heterogeneous autoimmune disease starting from dysregulation of the immune system and inflammation and followed by impaired angiogenesis, widespread vascular injury, and blood coagulation defects [22]. Persistently activated interstitial fibroblasts induce irreversible fibrosis of the skin and multiple internal organs. Except skin lesions, SSc is accompanied by cardiac defects (lesions in the coronary arteries, pericardium, and myocardium, and myocardial fibrosis) and renal failure (vascular lesions and fibrosis in the kidney, damage to renal glomeruli, and impaired glomerular filtration). Although the etiology of this life-threatening disorder is not completely understood, SSc-associated vasculopathy was demonstrated to result from hyperactivation of vascular endothelial cells due to genetic or environmental factors leading to dysregulated vascular remodeling. The central mechanism underlying tissue fibrosis is excessive deposition of extracellular matrix (ECM) proteins and impaired collagen homeostasis. Activated interstitial fibroblasts and immune cells synthesize excessive amounts of collagens (first of all types I, III, VI, and VII) and other ECM components such as fibronectin, tenascin-C (TNC), and alpha smooth muscle actin (αSMA), profibrotic cytokines including TGF-β and connective tissue growth factor CTGF (also known as CCN2), accompanied by reduced expression of ECM-degrading enzymes MMP (matrix metalloproteinases) 1 and 3 [23,24][23][24]. The causative relationship between transcription factors from the ETS family and pro-fibrotic processes was first established in the pioneering work of Czuwara-Ladykowska et al. [25], which demonstrated that Fli1 plays a crucial role in the regulation of collagen synthesis in dermal fibroblasts by competing with Ets-1, and the ratio of Fli1 to Ets-1 in the presence of co-regulatory proteins may control collagen production. Further in vitro studies have confirmed that FLI1 suppression induces SSc-like phenotypes in dermal fibroblasts, dermal dendritic cells, keratinocytes, endothelial cells, and macrophages, while in vivo findings have established that Fli1 deficiency is an important mediator of SSc-associated fibrosis [26,27,28][26][27][28]. The role of Fli1 as a natural negative inhibitor of collagen genes was supported by the fact that Fli1 protein levels were inversely proportional with COL1A1 mRNA and collagen 1 levels in the dermal fibroblasts of patients with SSc and in cultured fibroblast isolated from Fli1−/−, Fli1+/−, and Fli1+/+ mice embryos [29]. In the lesional and non-lesional skin of SSc patients, Fli1-positive fibroblasts are either consistently absent or only occasionally seen, and Fli1 immunoreactivity is significantly reduced in endothelial cells [29]. Fli1 is downregulated in dermal dendritic cells [30] and in the primary skin fibroblasts of SSc patients [31]. Furthermore, Fli1 has been demonstrated to play an important role in the regulation of other ECM proteins’ synthesis, including MMP-1 and CTGF [32,33][32][33]. Fli1 deficiency impairs the expression of various genes both in fibroblasts and endothelial cells and may represent a unique pathogenetic link between dermal fibrosis and peripheral vasculopathy in SSc. The critical role of Fli1 in the development of this disease was confirmed in the works on FLI1-mutated animals such as Fli1+/− mice [8] or that with a conditional deletion of FLI1 in endothelial cells (Fli1 CKO mice) [34]. Fli1-DNA binding. Under normal physiological conditions, Fli1 and Ets-1 form DNA–protein complexes with the sequences of the COL1A2 gene promoter. The suppressive interaction of Fli1with COL1 gene is functionally enhanced by co-regulating factors such as Sp1 and Sp3 [25]. Stable FLI1 transfection leads to dramatic suppression of COL1A2 mRNA and newly synthesized collagenous proteins by inhibiting COL1A2 promoter activity, while Ets-1 activates the COL1A2 promoter, which indicates that Fli1 inhibits the COL1A2 gene by displacing Ets-1. Recent studies have suggested that Fli1 also binds to promoters of genes encoding other proteins such as CCN2 [32], pro-angiogenic adipokine chemerin involved in inflammation, angiogenesis and energy metabolism [35], and one of the damage-associated molecular patterns (DAMPs) molecules S100A12 [36]. Fli1 signaling pathways. The transcriptional potential of Fli1 is strictly regulated by a balance between the activities of two cytokines—pro-fibrotic TGF-β, a well-established inductor of ECM proteins’ synthesis, and anti-fibrotic TNFα which prevents uncontrolled ECM production. TGF-β stimulation leads to activation of a few cellular targets (Figure 1, Table 1). Active PKC-δ is translocated to the nucleus and recruited to the COL1A2 promoter, where it directly phosphorylates Fli1 at threonine 312 [37]. Phosphorylation at this residue is necessary for interaction of Fli1 with p300/CREB-binding protein-associated factor (PCAF) resulting in acetylation of Fli1 at lysine 380 [38,39][38][39]. Acetylated Fli1 dissociates from the COL1A2 promoter and degrades, while decreased Fli1 transcription enhances the expression of COL genes. Another downstream effector of TGF-β is nonreceptor kinase c-Abl which activates the nuclear translocation of PKC-δ thus facilitating Fli1 acetylation and decreasing Fli1 repression of the COL1A2 gene [40]. Indeed, persistent stimulation of the c-Abl/PKC-δ/Fli1 cascade at least partially implicates in the fibrotic program in dermal fibroblasts obtained from patients with both SSc and its relative disease—localized scleroderma (LSc) [41]. Moreover, the c-Abl–PKC-δ–Fli1 pathway is activated by TGF-β-dependent stimulation of endothelin-1 [42].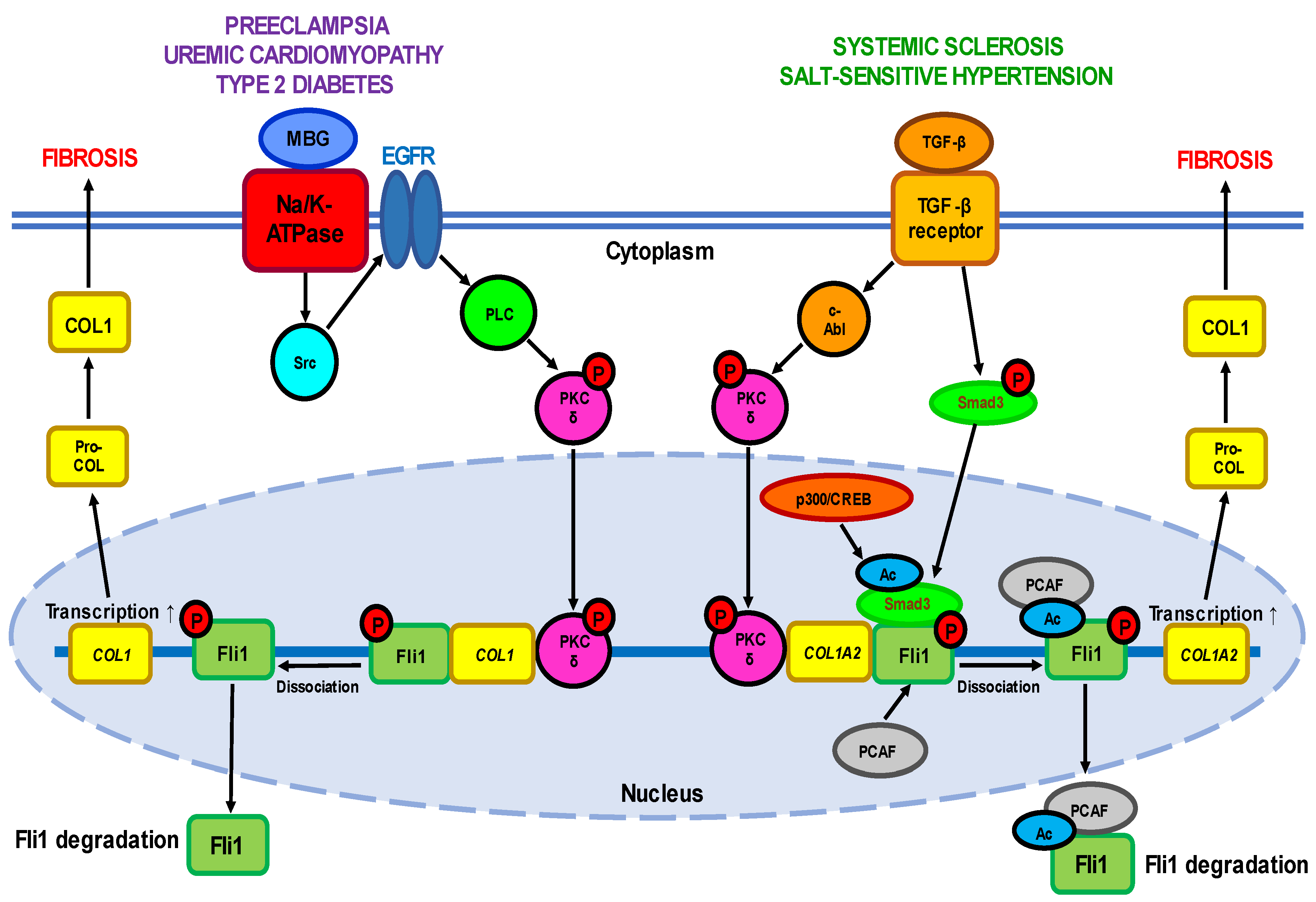
2.2. Fli1 in Uremic Cardiomyopathy
Uremic cardiomyopathy (UC) is a multifactorial pathological condition developed in the patients with chronic kidney disease (CKD) and is responsible for increased rates of heart failure and high mortality, particularly sudden cardiac death [64]. It is important that the risk of developing cardiovascular defects is even higher than that of kidney failure. UC is characterized by remodeling of cardiac tissues leading to hypertrophy of left ventricle and diastolic dysfunction, and by a myriad of metabolic maladaptations in myocardial cells including disturbed mitochondrial function, changes in substrate utilization and metabolic transporter function, and altered insulin response. The attempts to understand the cardiac perturbations accompanying CKD have revealed its link with abnormal Fli1 expression and increased collagen production as well (Figure 1). Early work has demonstrated that Fli1 transgenic mice developed a high incidence of immunological renal disease and ultimately died of renal failure caused by tubulointerstitial nephritis and immune-complex glomerulonephritis [65]. Later studies have strongly implicated the transcription factor Fli1 in the progressive cardiac fibrosis seen with experimental uremia. The works on rats that underwent partial nephrectomy have shown that many of the clinical features of experimental UC are accompanied by an increase in circulating concentrations of marinobufagenin (MBG), one of the cardiotonic steroids [66,67][66][67]. Moreover, MBG signaling through Na/K-ATPase is directly responsible for suppression of Fli1 and the stimulation of cardiac fibroblasts to produce increased amounts of collagen, thus triggering cardiac fibrosis seen with experimental renal failure [66,67][66][67]. These findings were confirmed by the fact that intraperitoneal administration of monoclonal antibodies to MBG restored Fli1 expression and reduced cardiac fibrosis [68]. Isolated cardiac fibroblasts cultured with 1 nM MBG in vitro increased procollagen-1 expression accompanied by synthesis of procollagen-1 mRNA and collagen translation [67]. The stimulation of fibroblasts with MBG was prevented by treatment with inhibitors of tyrosine phosphorylation, Src activation, EGFR transactivation, and N-acetyl cysteine. In general, the signaling pathway leading from MBG to increased collagen synthesis resembles that observed in other Fli1-deficiency-associated diseases such as SSc including translocation of PKC-δ to the nucleus where it stimulates Fli1 [16].2.3. Fli1 in Preeclampsia
Another example of pathology at least partially associated with aberrantly low Fli1 expression and the development of vascular fibrosis is preeclampsia (PE), a hypertensive complication of pregnancy adversely affecting both mothers and newborns and in some cases leading to maternal stroke and death [17,69,70][17][69][70]. PE is believed to depend on pre-existing cardiovascular abnormalities and is associated with a life-long risk of hypertension and coronary artery disease [70]. Among others, SARS-CoV-2 infection has recently been suggested to be linked with higher PE incidence, most probably due to inflammatory responses which can infect the placenta and impair its normal development [71]. An important role in PE belongs to maternal and placental vascular dysfunction, in particular abnormal remodeling of spiral arteries, endothelial damage, and an imbalance between the release of pro- and antiangiogenic factors (such as vascular endothelial growth factor A VEGFA, placental growth factor PlGF, soluble fms-like tyrosine kinase 1 sFLT-1, endoglin ENG, and soluble sENG) [72]. On the other hand, the progression of PE is closely related to enhanced synthesis of MBG and the development of cardiovascular fibrosis involving Fli1-dependent mechanisms [17,69][17][69]. Clinical studies have demonstrated that in umbilical arteries and placentas obtained from PE patients, the expression of Fli1 was significantly (4–9-fold) diminished, while the levels of pro-collagen and collagen-1 increased (2.5–3-fold) [73,74,75][73][74][75]. Similar results were obtained in the work on pregnant Sprague Dawley rats in which PE was induced experimentally by exposure to water with excessive NaCl amounts (1.8 %). The development of a PE-like phenotype in animals was accompanied by a seven-fold decrease in Fli1 expression and a four-fold increase in collagen-1 synthesis in thoracic aortas [76]. Finally, silencing of the FLI1 gene by 24 h incubation of the isolated segment of healthy human umbilical arteries with Fli1 siRNA not only suppressed the expression of Fli1 at the protein level, but also led to pronounced elevation of pro-collagen-1 and collagen-1 levels [77]. A causative relationship between MBG and Fli1 was confirmed in experiments in vitro on explants of umbilical arteries from healthy women. Incubation of the vascular fragments with 1-10 nM MBG for 24–48 h resulted in a considerable (4-5-fold) decrease in Fli1 levels and accumulation (2–3-fold) of pro-collagen-1 and collagen-1, thus mimicking the processes typical for PE and linked with vascular fibrosis [73,74,75][73][74][75]. In contrast, the degree of collagen-1 accumulation was diminished after the pre-treatment of arterial rings from normotensive pregnant women with monoclonal anti-MBG antibodies [74]. Application of canrenone, an active metabolite of spironolactone used in hypertensive therapy, fully restored Fli1 expression and partially suppressed collagen-1 synthesis in umbilical arteries from PE patients and in the vessels of healthy women incubated with MBG [75]. Moreover, short-term administration of anti-MBG antibodies to rats with experimental PE reversed Fli1 inhibition, although it had no effect on collagen-1 synthesis [76]. However, the underlying molecular pathways of PE in human or rat vessels were not associated with the stimulation of TGF-β and Smad proteins (Figure 1), although the protein level of cytoplasmic PKC-δ increased.2.4. Fli1 as a Common Causative Factor of Hypertension
Interestingly, the patients with preeclampsia [17], chronic renal failure [68[68][78],78], and type 2 diabetes [79] shared the same phenotype features, namely the development of cardiovascular fibrosis due to Fli1 deficiency. Researchers assume that the etiology and underlying molecular mechanisms of all these disorders are common and at least in part include enhanced levels of circulating MBG and inhibition of membrane enzyme Na/K-ATPase. The phenomena of MBG-induced fibrosis are sensitive to pharmacological modulation of Na/K-ATPase activity with anti-hypertensive agents [75]. The binding of MBG to Na/K-ATPase initiates the signal transduction that complexes with Src and epidermal growth factor receptor (EGFR) that leads to translocation of PKC-δ to the nucleus and Fli1 repression [16]. However, this process does not involve TGF-β—Smad proteins cascade, although TGF-β antagonist SB-431542 suppressed the stimulation of collagen production [67]. In contrast, Dahl salt-sensitive rats [80] and normotensive salt-loaded Sprague Dawley rats [81] react to a high-salt diet with fibrotic changes in myocardial and renal tissue which are accompanied by the activation of TGF-β [81]. Although the association of MBG and TGF-β described in these experimental models is clear, the causative relationship between them in salt sensitivity has not been explored in details. Interestingly, the crosstalk between Fli1 and TGF-β profibrotic signaling via PKC-δ has previously been described, for example, in type 2 diabetes during salt loading [79]. The development of diabetes is associated with profound inhibition of Fli1 in vascular tissues, while exposure of these rats to high salt levels activates TGF-β and suppresses the expression of Fli1 [79].| Expression | Effect | Experimental System | References | |
|---|---|---|---|---|
| Fli1 repressors | ||||
| TGF-β | ↑ | Activation of TGF-β receptors |
Human dermal fibroblasts; rat diabetes model; rat hypertension model |
[25][29][32][38][39][79][80][81] |
| c-Abl | ↑ | PKC-δ phosphorylation | Human normal and SSc dermal fibroblasts | [40][41][47][51] |
| PKC-δ | ↑ | Fli1 phosphorylation | Human normal and SSc dermal fibroblasts; FLI1-transfected HEK293T cells; FLI1 silencing in human UA |
[37][39][42][47][77] |
| Endothelin-1 | ↑ | Fli1 activation | Human normal and SSc dermal fibroblasts; BLM-treated mice |
[42] |
| PCAF | ↑ | Fli1 acetylation | Human dermal fibroblasts; FLI1-transfected HEK293T cells |
[38] |
| CXCL4 | ↑ | Vasculopathy | HUVECs | [51] |
| MBG | ↑ | PKC-δ activation; Fli1 suppression | Wild-type and FLI1-knockdown mice; rat CKD model; rat cardiac fibroblasts; human cardiac fibroblasts; human renal fibroblasts; FLI1-transfected renal fibroblasts; rat diabetes model; rat hypertension model; rat PE model |
[16][66][67][68][73][74][75][76][77][79][80][81] |
| Fli1-deficiency targets | ||||
| COL1 | ↑ | Collagen synthesis | FLI1-transfected human dermal fibroblasts; human normal and SSc dermal fibroblasts; murine Fli11−/−, Fli1+/− and Fli1+/+ fibroblasts; wild-type and FLI1-knockdown mice; rat CKD model; rat cardiac fibroblasts; human cardiac fibroblasts; human renal fibroblasts; FLI1-transfected renal fibroblasts; rat diabetes model; rat hypertension model; rat PE model; human PE UA; FLI1 silencing in human UA; human MBG-treated UA |
[8][16][25][29][32][38][66][67][68][73][74][75][76][77][79][80][81] |
| CTGF(CCN2) | ↑ | COL1A1 and COL1A2 upregulation; MMP-1 downregulation | FLI1-transfected human dermal fibroblasts; human SSc fibroblasts |
[32][33] |
| MMP-1 | ↓ | Increased production of EMC components | FLI1-transfected human dermal fibroblasts; human SSc fibroblasts |
[32][33] |
| Fibronectin | ↑ | Fibrosis | Human CD14+ monocytes and CD14+ macrophages | [23] |
| Chemerin | ↑ | Impaired angiogenesis | Human SSc dermal vessels and HDMECs; murine Fli1+/− dermal blood vessels; |
[35] |
| S100A12 | ↑ | Skin sclerosis | Human SSc skin; | [36] |
| RALDH1 | ↓ | Fibrosis | Human dermal dendritic cells; BLM-treated Fli1+/− mice |
[30] |
| Cathepsin B | ↑ ↓ |
Vasculopathy Increased production of EMC components |
FLI1 silencing in HDMECs; Fli1+/− mice; human dcSSc dermal fibroblasts, early dcSSc dermal fibroblasts |
[43] |
| Cathepsin L | ↑ ↓ |
Vasculopathy Increased production of EMC components |
FLI1 silencing in HDMECs; human SS dermal vessels and skin; FLI1-knockout mice; skin of BLM-treated mice |
[45] |
| Cathepsin V | ↓ | Increased production of EMC components | FLI1 silencing in HDMECs; human dcSSc dermal fibroblasts, microvascular ECs, dcSSc and lcSSc skin keratinocytes |
[44] |
| PGRN | ↑ | Inflammation; skin sclerosis; fibrosis |
Human SSc dermal fibroblasts; Fli-1+/− mice; BLM-treated mice; human LSc skin lesions |
[46][47] |
| TNF-α | ↑ | Inflammation; fibrosis |
Human LSc skin lesions; | [47] |
| CXCL5 | ↓ | Vasculopathy | Human dcSSc dermal blood vessels; FLI1 silencing in HDMECs; dermal vessels of Fli1 knockout mice |
[48] |
| CXCL6 | ↑ | Tissues fibrosis; vasculopathy |
Human SSc dermal fibroblasts; FLI1 silencing in HDMECs |
[49] |
| CCL20 | ↑ | Fibrosis | Human early dcSSc | [52] |
| CCL6 | ↑ | Vasculopathy | Human SSc dermal vessels; FLI1 silencing in HDMECs |
[52] |
| EPCR | ↓ | Impaired vascular homeostasis | Human SSc dermal vessels; Fli1+/− mice; FLI1 silencing in HDMECs |
[53] |
| VE-cadherin | ↓ | Impaired vascular homeostasis | MDMECs of Fli1 CKO mice; MDMECs of Fli1+/− mice; FLI1 silencing in HDMECs |
[34] |
| PECAM-1 | ↓ | Impaired vascular homeostasis | MDMECs of Fli1 CKO mice; MDMECs of Fli1+/− mice; FLI1 silencing in HDMECs |
[34] |
| MMP-9 | ↓ | Impaired vascular homeostasis | MDMECs of Fli1 CKO mice; MDMECs of Fli1+/− mice; FLI1 silencing in HDMECs |
[34] |
| PDGF-B | ↓ | Impaired vascular homeostasis | MDMECs of Fli1 CKO mice; MDMECs of Fli1+/− mice; FLI1 silencing in HDMECs |
[34] |
| S1P1 | ↓ | Impaired vascular homeostasis | MDMECs of Fli1 CKO mice; MDMECs of Fli1+/− mice; FLI1 silencing in HDMECs |
[34] |
| Adipsin | ↑ | Vascular hypertension | Human SSc dermal vessels; FLI1 silencing in HDMECs |
[54] |
| Genetic and epigenetic factors | ||||
| (GA)n alleles | ↑ | Increased susceptibility to SSc | Human SSc peripheral blood | [56] |
| Acetylation of histones H3 and H4 in the FLI1 gene promoter | ↓ | FLI1 suppression; increased collagen synthesis | Human skin, normal and dcSSc fibroblasts | [57] |
| HDAC-1 and 6 | ↑ | FLI1 suppression | Human skin, normal and dcSSc fibroblasts | [57] |
| MBD-1 and 2, MeCP2 | ↑ | DNA methylation | Human skin, normal and dcSSc fibroblasts | [57] |
| Methylation of CpG islands in the FLI1 promoter | ↑ | FLI1 suppression; increased Collagen synthesis | Human skin, normal and dcSSc fibroblasts | [57] |
| DNMT1 | ↑ | FLI1 suppression | Human skin, normal and dcSSc fibroblasts | [33][57] |
| COL23A1, COL4A2 methylation | ↓ | Increased collagen synthesis | Human dcSSc and lcSSc dermal fibroblasts | [58] |
| ITGA9 methylation | ↓ | TGF-β upregulation | Human dcSSc and lcSSc dermal fibroblasts | [58] |
| ADAM12 methylation | ↓ | TGF-β upregulation | Human dcSSc and lcSSc dermal fibroblasts | [58] |
| miRNA-26a | ↑ | FLI1 suppression | Primary SSc skin fibroblasts | [31] |
| miRNA-21 | ↑ | TGF-β upregulation; collagen synthesis |
Human dcSSc and TGF-β treated normal fibroblasts | [59] |
| miRNA-29a | ↓ | TGF-β upregulation; collagen synthesis |
Human dcSSc and TGF-β treated normal fibroblasts | [59] |
| Expression | Effect | Experimental System | References | |
|---|---|---|---|---|
| Fli1 repressors | ||||
| TGF-β | ↑ | Activation of TGF-β receptors |
Human dermal fibroblasts; rat diabetes model; rat hypertension model |
[25,29,32,38,39,79,80,81] |
| c-Abl | ↑ | PKC-δ phosphorylation | Human normal and SSc dermal fibroblasts | [40,41,47,51] |
| PKC-δ | ↑ | Fli1 phosphorylation | Human normal and SSc dermal fibroblasts; FLI1-transfected HEK293T cells; FLI1 silencing in human UA |
[37,39,42,47,77] |
| Endothelin-1 | ↑ | Fli1 activation | Human normal and SSc dermal fibroblasts; BLM-treated mice |
[42] |
| PCAF | ↑ | Fli1 acetylation | Human dermal fibroblasts; FLI1-transfected HEK293T cells |
[38] |
| CXCL4 | ↑ | Vasculopathy | HUVECs | [51] |
| MBG | ↑ | PKC-δ activation; Fli1 suppression | Wild-type and FLI1-knockdown mice; rat CKD model; rat cardiac fibroblasts; human cardiac fibroblasts; human renal fibroblasts; FLI1-transfected renal fibroblasts; rat diabetes model; rat hypertension model; rat PE model |
[16,66,67,68,73,74,75,76,77,79,80,81] |
| Fli1-deficiency targets | ||||
| COL1 | ↑ | Collagen synthesis | FLI1-transfected human dermal fibroblasts; human normal and SSc dermal fibroblasts; murine Fli11−/−, Fli1+/− and Fli1+/+ fibroblasts; wild-type and FLI1-knockdown mice; rat CKD model; rat cardiac fibroblasts; human cardiac fibroblasts; human renal fibroblasts; FLI1-transfected renal fibroblasts; rat diabetes model; rat hypertension model; rat PE model; human PE UA; FLI1 silencing in human UA; human MBG-treated UA |
[8,16,25,29,32,38,66,67,68,73,74,75,76,77,79,80,81] |
| CTGF(CCN2) | ↑ | COL1A1 and COL1A2 upregulation; MMP-1 downregulation | FLI1-transfected human dermal fibroblasts; human SSc fibroblasts |
[32,33] |
| MMP-1 | ↓ | Increased production of EMC components | FLI1-transfected human dermal fibroblasts; human SSc fibroblasts |
[32,33] |
| Fibronectin | ↑ | Fibrosis | Human CD14+ monocytes and CD14+ macrophages | [23] |
| Chemerin | ↑ | Impaired angiogenesis | Human SSc dermal vessels and HDMECs; murine Fli1+/− dermal blood vessels; |
[35] |
| S100A12 | ↑ | Skin sclerosis | Human SSc skin; | [36] |
| RALDH1 | ↓ | Fibrosis | Human dermal dendritic cells; BLM-treated Fli1+/− mice |
[30] |
| Cathepsin B | ↑ ↓ |
Vasculopathy Increased production of EMC components |
FLI1 silencing in HDMECs; Fli1+/− mice; human dcSSc dermal fibroblasts, early dcSSc dermal fibroblasts |
[43] |
| Cathepsin L | ↑ ↓ |
Vasculopathy Increased production of EMC components |
FLI1 silencing in HDMECs; human SS dermal vessels and skin; FLI1-knockout mice; skin of BLM-treated mice |
[45] |
| Cathepsin V | ↓ | Increased production of EMC components | FLI1 silencing in HDMECs; human dcSSc dermal fibroblasts, microvascular ECs, dcSSc and lcSSc skin keratinocytes |
[44] |
| PGRN | ↑ | Inflammation; skin sclerosis; fibrosis |
Human SSc dermal fibroblasts; Fli-1+/− mice; BLM-treated mice; human LSc skin lesions |
[46,47] |
| TNF-α | ↑ | Inflammation; fibrosis |
Human LSc skin lesions; | [47] |
| CXCL5 | ↓ | Vasculopathy | Human dcSSc dermal blood vessels; FLI1 silencing in HDMECs; dermal vessels of Fli1 knockout mice |
[48] |
| CXCL6 | ↑ | Tissues fibrosis; vasculopathy |
Human SSc dermal fibroblasts; FLI1 silencing in HDMECs |
[49] |
| CCL20 | ↑ | Fibrosis | Human early dcSSc | [52] |
| CCL6 | ↑ | Vasculopathy | Human SSc dermal vessels; FLI1 silencing in HDMECs |
[52] |
| EPCR | ↓ | Impaired vascular homeostasis | Human SSc dermal vessels; Fli1+/− mice; FLI1 silencing in HDMECs |
[53] |
| VE-cadherin | ↓ | Impaired vascular homeostasis | MDMECs of Fli1 CKO mice; MDMECs of Fli1+/− mice; FLI1 silencing in HDMECs |
[34] |
| PECAM-1 | ↓ | Impaired vascular homeostasis | MDMECs of Fli1 CKO mice; MDMECs of Fli1+/− mice; FLI1 silencing in HDMECs |
[34] |
| MMP-9 | ↓ | Impaired vascular homeostasis | MDMECs of Fli1 CKO mice; MDMECs of Fli1+/− mice; FLI1 silencing in HDMECs |
[34] |
| PDGF-B | ↓ | Impaired vascular homeostasis | MDMECs of Fli1 CKO mice; MDMECs of Fli1+/− mice; FLI1 silencing in HDMECs |
[34] |
| S1P1 | ↓ | Impaired vascular homeostasis | MDMECs of Fli1 CKO mice; MDMECs of Fli1+/− mice; FLI1 silencing in HDMECs |
[34] |
| Adipsin | ↑ | Vascular hypertension | Human SSc dermal vessels; FLI1 silencing in HDMECs |
[54] |
| Genetic and epigenetic factors | ||||
| (GA)n alleles | ↑ | Increased susceptibility to SSc | Human SSc peripheral blood | [56] |
| Acetylation of histones H3 and H4 in the FLI1 gene promoter | ↓ | FLI1 suppression; increased collagen synthesis | Human skin, normal and dcSSc fibroblasts | [57] |
| HDAC-1 and 6 | ↑ | FLI1 suppression | Human skin, normal and dcSSc fibroblasts | [57] |
| MBD-1 and 2, MeCP2 | ↑ | DNA methylation | Human skin, normal and dcSSc fibroblasts | [57] |
| Methylation of CpG islands in the FLI1 promoter | ↑ | FLI1 suppression; increased Collagen synthesis | Human skin, normal and dcSSc fibroblasts | [57] |
| DNMT1 | ↑ | FLI1 suppression | Human skin, normal and dcSSc fibroblasts | [33,57] |
| COL23A1, COL4A2 methylation | ↓ | Increased collagen synthesis | Human dcSSc and lcSSc dermal fibroblasts | [58] |
| ITGA9 methylation | ↓ | TGF-β upregulation | Human dcSSc and lcSSc dermal fibroblasts | [58] |
| ADAM12 methylation | ↓ | TGF-β upregulation | Human dcSSc and lcSSc dermal fibroblasts | [58] |
| miRNA-26a | ↑ | FLI1 suppression | Primary SSc skin fibroblasts | [31] |
| miRNA-21 | ↑ | TGF-β upregulation; collagen synthesis |
Human dcSSc and TGF-β treated normal fibroblasts | [59] |
| miRNA-29a | ↓ | TGF-β upregulation; collagen synthesis |
Human dcSSc and TGF-β treated normal fibroblasts | [59] |
References
- Ben-David, Y.; Giddens, E.B.; Bernstein, A. Identification and mapping of a common proviral integration site Fli-1 in erythroleukemia cells induced by Friend murine leukemia virus. Proc. Natl. Acad. Sci. USA 1990, 87, 1332–1336.
- Ben-David, Y.; Giddens, E.B.; Letwin, K.; Bernstein, A. Erythroleukemia induction by Friend murine leukemia virus: Insertional activation of a new member of the ets gene family, Fli-1, closely linked to c-ets-1. Genes Dev. 1991, 5, 908–918.
- He, Y.S.; Yang, X.K.; Hu, Y.Q.; Xiang, K.; Pan, H.F. Emerging role of Fli1 in autoimmune diseases. Int. Immunopharmacol. 2021, 90, 107127.
- Ben-David, Y.; Babu Gajendran, B.; Klarke, M.; Sample, K.M.; Zacksenhaus, E. Current insights into the role of Fli-1 in hematopoiesis and malignant transformation. Cell. Mol. Life Sci. 2022, 79, 163.
- Sato, Y. Role of ETS family transcription factors in vascular development and angiogenesis. Cell Struct. Funct. 2001, 26, 19–24.
- Ferdous, A.; Singh, S.; Luo, Y.; Abedin, M.J.; Jiang, N.; Perry, C.E.; Evers, B.M.; Gillette, T.G.; Kyba, M.; Trojanowska, M.; et al. Fli1 Promotes Vascular Morphogenesis by Regulating Endothelial Potential of Multipotent Myogenic Progenitors. Circ. Res. 2021, 129, 949–964.
- Spyropoulos, D.D.; Pharr, P.N.; Lavenburg, K.R.; Jackers, P.; Papas, T.S.; Ogawa, M.; Watson, D.K. Hemorrhage, impaired hematopoiesis, and lethality in mouse embryos carrying a targeted disruption of the Fli1 transcription factor. Mol. Cell. Biol. 2000, 20, 5643–5652.
- Asano, Y.J. What can we learn from Fli1-deficient mice, new animal models of systemic sclerosis? Scleroderma Relat. Disord. 2018, 3, 6–13.
- Pimanda, J.E.; Chan, W.Y.; Donaldson, I.J.; Bowen, M.; Green, A.R.; Göttgens, B. Endoglin expression in the endothelium is regulated by Fli-1, Erg, and Elf-1 acting on the promoter and a −8-kb enhancer. Blood 2006, 107, 4737–4745.
- Le Bras, A.; Samson, C.; Trentini, M.; Caetano, B.; Lelievre, E.; Mattot, V.; Beermann, F.; Soncin, F. VE-statin/egfl7 expression in endothelial cells is regulated by a distal enhancer and a proximal promoter under the direct control of Erg and GATA-2. PLoS ONE 2010, 5, e12156.
- Soncin, F.; Mattot, V.; Lionneton, F.; Spruyt, N.; Lepretre, F.; Begue, A.; Stehelin, D. VE-statin, an endothelial repressor of smooth muscle cell migration. EMBO J. 2003, 22, 5700–5711.
- Lelièvre, E.; Lionneton, F.; Mattot, V.; Spruyt, N.; Soncin, F. Ets-1 regulates fli-1 expression in endothelial cells. Identification of ETS binding sites in the fli-1 gene promoter. J. Biol. Chem. 2002, 277, 25143–25151.
- Li, L.; Yu, J.; Cheng, S.; Peng, Z.; Luo, H. Transcription factor Fli-1 as a new target for antitumor drug development. Int. J. Biol. Macromol. 2022, 209 Pt A, 1155–1168.
- Gargallo, P.; Yáñez, Y.; Juan, A.; Segura, V.; Balaguer, J.; Torres, B.; Oltra, S.; Castel, V.; Cañete, A. Review: Ewing Sarcoma Predisposition. Pathol. Oncol. Res. 2020, 26, 2057–2066.
- Qian, C.; Li, D.; Chen, Y. ETS factors in prostate cancer. Cancer Lett. 2022, 530, 181–189.
- Elkareh, J.; Periyasamy, S.M.; Shidyak, A.; Vetteth, S.; Schroeder, J.; Raju, V.; Hariri, I.M.; El-Okdi, N.; Gupta, S.; Fedorova, L.; et al. Marinobufagenin induces increases in procollagen expression in a process involving protein kinase C and Fli-1: Implications for uremic cardiomyopathy. Am. J. Physiol. Renal Physiol. 2009, 296, F1219–F1226.
- Agalakova, N.I.; Kolodkin, N.I.; Adair, C.D.; Trashkov, A.P.; Bagrov, A.Y. Preeclampsia: Cardiotonic Steroids, Fibrosis, Fli1 and Hint to Carcinogenesis. Int. J. Mol. Sci. 2021, 22, 1941.
- Toyama, T.; Asano, Y.; Miyagawa, T.; Nakamura, K.; Hirabayashi, M.; Yamashita, T.; Saigusa, R.; Miura, S.; Ichimura, Y.; Takahashi, T.; et al. The impact of transcription factor Fli1 deficiency on the regulation of angiogenesis. Exp. Dermatol. 2017, 26, 912–918.
- Asano, Y. Epigenetic suppression of Fli1, a potential predisposing factor in the pathogenesis of systemic sclerosis. Int. J. Biochem. Cell Biol. 2015, 67, 86–91.
- Fioretto, B.S.; Rosa, I.; Romano, E.; Wang, Y.; Guiducci, S.; Zhang, G.; Manetti, M.; Matucci-Cerinic, M. The contribution of epigenetics to the pathogenesis and gender dimorphism of systemic sclerosis: A comprehensive overview. Ther. Adv. Musculoskelet. Dis. 2020, 12, 1759720X20918456.
- Yu, J.; Tang, R.; Ding, K. Epigenetic Modifications in the Pathogenesis of Systemic Sclerosis. Int. J. Gen. Med. 2022, 15, 3155–3166.
- Kowalska-Kępczyńska, A.J. Systemic Scleroderma-Definition, Clinical Picture and Laboratory Diagnostics. Clin. Med. 2022, 11, 2299.
- Rudnik, M.; Hukara, A.; Kocherova, I.; Jordan, S.; Schniering, J.; Milleret, V.; Ehrbar, M.; Klingel, K.; Feghali-Bostwick, C.; Distler, O.; et al. Elevated Fibronectin Levels in Profibrotic CD14+ Monocytes and CD14+ Macrophages in Systemic Sclerosis. Front. Immunol. 2021, 12, 642891.
- Bhattacharyya, S.; Midwood, K.S.; Varga, J. Tenascin-C in fibrosis in multiple organs: Translational implications. Semin. Cell Dev. Biol. 2022, 128, 130–136.
- Czuwara-Ladykowska, J.; Shirasaki, F.; Jackers, P.; Watson, D.K.; Trojanowska, M.J. Fli-1 inhibits collagen type I production in dermal fibroblasts via an Sp1-dependent pathway. Biol. Chem. 2001, 276, 20839–20848.
- Manetti, M. Fli1 deficiency and beyond: A unique pathway linking peripheral vasculopathy and dermal fibrosis in systemic sclerosis. Exp. Dermatol. 2015, 24, 256–257.
- Takahashi, T.; Asano, Y.; Sugawara, K.; Yamashita, T.; Nakamura, K.; Saigusa, R.; Ichimura, Y.; Toyama, T.; Taniguchi, T.; Akamata, K.; et al. Epithelial Fli1 deficiency drives systemic autoimmunity and fibrosis: Possible roles in scleroderma. J. Exp. Med. 2017, 214, 1129–1151.
- Asano, Y.J. The Pathogenesis of Systemic Sclerosis: An Understanding Based on a Common Pathologic Cascade across Multiple Organs and Additional Organ-Specific Pathologies. Clin. Med. 2020, 9, 2687.
- Kubo, M.; Czuwara-Ladykowska, J.; Moussa, O.; Markiewicz, M.; Smith, E.; Silver, R.M.; Jablonska, S.; Blaszczyk, M.; Watson, D.K.; Trojanowska, M. Persistent down-regulation of Fli1, a suppressor of collagen transcription, in fibrotic scleroderma skin. Am. J. Pathol. 2003, 163, 571–581.
- Miura, S.; Watanabe, Y.; Saigusa, R.; Yamashita, T.; Nakamura, K.; Hirabayashi, M.; Miyagawa, T.; Yoshizaki, A.; Trojanowska, M.; Sato, S.; et al. Fli1 deficiency suppresses RALDH1 activity of dermal dendritic cells and related induction of regulatory T cells: A possible role in scleroderma. Arthritis Res. Ther. 2021, 23, 137.
- Cheng, Z.; Zhang, J.; Deng, W.; Lin, S.; Li, D.; Zhu, K.; Qi, Q. Bushen Yijing Decoction (BSYJ) exerts an anti-systemic sclerosis effect via regulating MicroRNA-26a/FLI1 axis. Bioengineered 2021, 12, 1212–1225.
- Nakerakanti, S.S.; Kapanadze, B.; Yamasaki, M.; Markiewicz, M.; Trojanowska, M. Fli1 and Ets1 have distinct roles in connective tissue growth factor/CCN2 gene regulation and induction of the profibrotic gene program. J. Biol. Chem. 2006, 281, 25259–25269.
- Bujor, A.M.; Haines, P.; Padilla, C.; Christmann, R.B.; Junie, M.; Sampaio-Barros, P.D.; Lafyatis, R.; Trojanowska, M. Ciprofloxacin has antifibrotic effects in scleroderma fibroblasts via downregulation of Dnmt1 and upregulation of Fli1. Int. J. Mol. Med. 2012, 30, 1473–1480.
- Asano, Y.; Stawski, L.; Hant, F.; Highland, K.; Silver, R.; Szalai, G.; Watson, D.K.; Trojanowska, M. Endothelial Fli1 deficiency impairs vascular homeostasis: A role in scleroderma vasculopathy. Am. J. Pathol. 2010, 176, 1983–1998.
- Akamata, K.; Asano, Y.; Taniguchi, T.; Yamashita, T.; Saigusa, R.; Nakamura, K.; Noda, S.; Aozasa, N.; Toyama, T.; Takahashi, T.; et al. Increased expression of chemerin in endothelial cells due to Fli1 deficiency may contribute to the development of digital ulcers in systemic sclerosis. Rheumatology 2015, 54, 1308–1316.
- Omatsu, J.; Saigusa, R.; Miyagawa, T.; Fukui, Y.; Toyama, S.; Awaji, K.; Ikawa, T.; Norimatsu, Y.; Yoshizaki, A.; Sato, S.; et al. Serum S100A12 levels: Possible association with skin sclerosis and interstitial lung disease in systemic sclerosis. Exp. Dermatol. 2021, 30, 409–415.
- Jinnin, M.; Ihn, H.; Yamane, K.; Mimura, Y.; Asano, Y.; Tamaki, K. Alpha2(I) collagen gene regulation by protein kinase C signaling in human dermal fibroblasts. Nucleic Acids Res. 2005, 33, 1337–1351.
- Asano, Y.; Czuwara, J.; Trojanowska, M. Transforming growth factor-beta regulates DNA binding activity of transcription factor Fli1 by p300/CREB-binding protein-associated factor-dependent acetylation. J. Biol. Chem. 2007, 282, 34672–34683.
- Asano, Y.; Trojanowska, M. Phosphorylation of Fli1 at threonine 312 by protein kinase C delta promotes its interaction with p300⁄CREB-binding protein-associated factor and subsequent acetylation in response to transforming growth factor beta. Mol. Cell. Biol. 2009, 29, 1882–1894.
- Bujor, A.M.; Asano, Y.; Haines, P.; Lafyatis, R.; Trojanowska, M. The c-Abl tyrosine kinase controls protein kinase Cδ-induced Fli-1 phosphorylation in human dermal fibroblasts. Arthritis Rheum. 2011, 63, 1729–1737.
- Noda, S.; Asano, Y.; Akamata, K.; Aozasa, N.; Taniguchi, T.; Takahashi, T.; Ichimura, Y.; Toyama, T.; Sumida, H.; Yanaba, K.; et al. Constitutive activation of c-Abl/protein kinase C-δ/Fli1 pathway in dermal fibroblasts derived from patients with localized scleroderma. Br. J. Dermatol. 2012, 167, 1098–1105.
- Akamata, K.; Asano, Y.; Aozasa, N.; Noda, S.; Taniguchi, T.; Takahashi, T.; Ichimura, Y.; Toyama, T.; Sato, S. Bosentan reverses the pro-fibrotic phenotype of systemic sclerosis dermal fibroblasts via increasing DNA binding ability of transcription factor Fli1. Arthritis Res. Ther. 2014, 16, R86.
- Noda, S.; Asano, Y.; Akamata, K.; Aozasa, N.; Taniguchi, T.; Takahashi, T.; Ichimura, Y.; Toyama, T.; Sumida, H.; Yanaba, K.; et al. A possible contribution of altered cathepsin B expression to the development of skin sclerosis and vasculopathy in systemic sclerosis. PLoS ONE 2012, 7, e32272.
- Noda, S.; Asano, Y.; Takahashi, T.; Akamata, K.; Aozasa, N.; Taniguchi, T.; Ichimura, Y.; Toyama, T.; Sumida, H.; Kuwano, Y.; et al. Decreased cathepsin V expression due to Fli1 deficiency contributes to the development of dermal fibrosis and proliferative vasculopathy in systemic sclerosis. Rheumatology 2013, 52, 790–799.
- Yamashita, T.; Asano, Y.; Taniguchi, T.; Nakamura, K.; Saigusa, R.; Takahashi, T.; Ichimura, Y.; Toyama, T.; Yoshizaki, A.; Miyagaki, T.; et al. A potential contribution of altered cathepsin L expression to the development of dermal fibrosis and vasculopathy in systemic sclerosis. Exp. Dermatol. 2016, 25, 287–292.
- Ichimura, Y.; Asano, Y.; Akamata, K.; Noda, S.; Taniguchi, T.; Takahashi, T.; Toyama, T.; Tada, Y.; Sugaya, M.; Sato, S.; et al. Progranulin Overproduction Due to Fli-1 Deficiency Contributes to the Resistance of Dermal Fibroblasts to Tumor Necrosis Factor in Systemic Sclerosis. Arthritis Rheumatol. 2015, 67, 3245–3255.
- Miyagawa, T.; Ichimura, Y.; Nakamura, K.; Hirabayashi, M.; Yamashita, T.; Saigusa, R.; Miura, S.; Takahashi, T.; Toyama, T.; Taniguchi, T.; et al. Progranulin overproduction due to constitutively activated c-Abl/PKC-δ/Fli1 pathway contributes to the resistance of dermal fibroblasts to the anti-fibrotic effect of tumor necrosis factor-α in localized scleroderma. Dermatol. Sci. 2018, 92, 207–214.
- Ichimura, Y.; Asano, Y.; Akamata, K.; Takahashi, T.; Noda, S.; Taniguchi, T.; Toyama, T.; Aozasa, N.; Sumida, H.; Kuwano, Y.; et al. Fli1 deficiency contributes to the suppression of endothelial CXCL5 expression in systemic sclerosis. Arch. Dermatol. Res. 2014, 306, 331–338.
- Taniguchi, T.; Asano, Y.; Nakamura, K.; Yamashita, T.; Saigusa, R.; Ichimura, Y.; Takahashi, T.; Toyama, T.; Yoshizaki, A.; Sato, S. Fli1 Deficiency Induces CXCL6 Expression in Dermal Fibroblasts and Endothelial Cells, Contributing to the Development of Fibrosis and Vasculopathy in Systemic Sclerosis. J. Rheumatol. 2017, 44, 1198–1205.
- Taniguchi, T.; Miyagawa, T.; Toyama, S.; Yamashita, T.; Nakamura, K.; Saigusa, R.; Ichimura, Y.; Takahashi, T.; Toyama, T.; Yoshizaki, A.; et al. CXCL13 produced by macrophages due to Fli1 deficiency may contribute to the development of tissue fibrosis, vasculopathy and immune activation in systemic sclerosis. Exp. Dermatol. 2018, 27, 1030–1037.
- Jiang, Z.; Chen, C.; Yang, S.; He, H.; Zhu, X.; Liang, M. Contribution to the peripheral vasculopathy and endothelial cell dysfunction by CXCL4 in Systemic Sclerosis. J. Dermatol. Sci. 2021, 104, 63–73.
- Ikawa, T.; Miyagawa, T.; Fukui, Y.; Toyama, S.; Omatsu, J.; Awaji, K.; Norimatsu, Y.; Watanabe, Y.; Yoshizaki, A.; Sato, S.; et al. Endothelial CCR6 expression due to FLI1 deficiency contributes to vasculopathy associated with systemic sclerosis. Arthritis Res. Ther. 2021, 23, 283.
- Saigusa, R.; Asano, Y.; Yamashita, T.; Taniguchi, T.; Takahashi, T.; Ichimura, Y.; Toyama, T.; Yoshizaki, A.; Miyagaki, T.; Sugaya, M.; et al. Fli1 deficiency contributes to the downregulation of endothelial protein C receptor in systemic sclerosis: A possible role in prothrombotic conditions. Br. J. Dermatol. 2016, 174, 338–347.
- Miyagawa, T.; Taniguchi, T.; Saigusa, R.; Fukayama, M.; Takahashi, T.; Yamashita, T.; Hirabayashi, M.; Miura, S.; Nakamura, K.; Yoshizaki, A.; et al. Fli1 deficiency induces endothelial adipsin expression, contributing to the onset of pulmonary arterial hypertension in systemic sclerosis. Rheumatology 2020, 59, 2005–2015.
- Mayes, M.D.; Trojanowska, M. Genetic factors in systemic sclerosis. Arthritis Res. Ther. 2007, 9, S5.
- Yamashita, K.; Kawasaki, A.; Matsushita, T.; Furukawa, H.; Kondo, Y.; Okiyama, N.; Nagaoka, S.; Shimada, K.; Sugii, S.; Katayama, M.; et al. Association of functional (GA)n microsatellite polymorphism in the FLI1 gene with susceptibility to human systemic sclerosis. Rheumatology 2020, 59, 3553–3562.
- Wang, Y.; Fan, P.S.; Kahaleh, B. Association between enhanced type I collagen expression and epigenetic repression of the FLI1 gene in scleroderma fibroblasts. Arthritis Rheum. 2006, 54, 2271–2279.
- Altorok, N.; Tsou, P.S.; Coit, P.; Khanna, D.; Sawalha, A.H. Genome-wide DNA methylation analysis in dermal fibroblasts from patients with diffuse and limited systemic sclerosis reveals common and subset-specific DNA methylation aberrancies. Ann. Rheum. Dis. 2015, 74, 1612–1620.
- Jafarinejad-Farsangi, S.; Gharibdoost, F.; Farazmand, A.; Kavosi, H.; Jamshidi, A.; Karimizadeh, E.; Noorbakhsh, F.; Mahmoudi, M. MicroRNA-21 and microRNA-29a modulate the expression of collagen in dermal fibroblasts of patients with systemic sclerosis. Autoimmunity 2019, 52, 108–116.
- Maurer, B.; Stanczyk, J.; Jüngel, A.; Akhmetshina, A.; Trenkmann, M.; Brock, M.; Kowal-Bielecka, O.; Gay, R.E.; Michel, B.A.; Distler, J.H.; et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum. 2010, 62, 1733–1743.
- Makino, K.; Jinnin, M.; Hirano, A.; Yamane, K.; Eto, M.; Kusano, T.; Honda, N.; Kajihara, I.; Makino, T.; Sakai, K.; et al. The downregulation of miR-let-7a contributes to the excessive expression of type I collagen in systemic and localized scleroderma. J. Immunol. 2013, 190, 3905–3915.
- Makino, T.; Jinnin, M.; Etoh, M.; Yamane, K.; Kajihara, I.; IMakino, H.; Ichihara, A.; Igata, T.; Sakai, K.; Fukushima, S.; et al. Downregulation of microRNA-196a in the sera and involved skin of localized scleroderma patients. Eur. J. Dermatol. 2014, 24, 470–476.
- O’Reilly, S.; Ciechomska, M.; Fullard, N.; Przyborski, S.; van Laar, J.M. IL-13 mediates collagen deposition via STAT6 and microRNA-135b: A role for epigenetics. Sci. Rep. 2016, 6, 25066.
- Garikapati, K.; Goh, D.; Khanna, S.; Echampati, K. Uraemic Cardiomyopathy: A Review of Current Literature. Clin. Med. Insights Cardiol. 2021, 15, 1179546821998347.
- Zhang, L.; Eddy, A.; Teng, Y.T.; Fritzler, M.; Kluppel, M.; Melet, F.; Bernstein, A. An immunological renal disease in transgenic mice that overexpress Fli-1, a member of the ets family of transcription factor genes. Mol. Cell. Biol. 1995, 15, 6961–6970.
- Kennedy, D.J.; Vetteth, S.; Periyasamy, S.M.; Kanj, M.; Fedorova, L.; Khouri, S.; Kahaleh, M.B.; Xie, Z.; Malhotra, D.; Kolodkin, N.I.; et al. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension 2006, 47, 488–495.
- Elkareh, J.; Kennedy, D.J.; Yashaswi, B.; Vetteth, S.; Shidyak, A.; Kim, E.G.; Smaili, S.; Periyasamy, S.M.; Hariri, I.M.; Fedorova, L.; et al. Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension 2007, 49, 215–224.
- Haller, S.T.; Kennedy, D.J.; Shidyak, A.; Budny, G.V.; Malhotra, D.; Fedorova, O.V.; Shapiro, J.I.; Bagrov, A.Y. Monoclonal antibody against marinobufagenin reverses cardiac fibrosis in rats with chronic renal failure. Am. J. Hypertens. 2012, 25, 690–696.
- Reznik, V.A.; Kashkin, V.A.; Agalakova, N.I.; Adair, C.D.; Bagrov, A.Y. Endogenous Bufadienolides, Fibrosis and Preeclampsia. Cardiol. Res. Pract. 2019, 2019, 5019287.
- Dennehy, N.; Lees, C. Preeclampsia: Maternal cardiovascular function and optimising outcomes. Early Hum. Dev. 2022, 174, 105669.
- Tossetta, G.; Fantone, S.; Muti, N.D.; Balercia, G.; Ciavattini, A.; Giannubilo, S.R.; Marzioni, D. Preeclampsia and severe acute respiratory syndrome coronavirus 2 infection: A systematic review. J. Hypertens. 2022, 40, 629–1638.
- Opichka, M.A.; Rappelt, M.W.; Gutterman, D.D.; Grobe, J.L.; McIntosh, J.J. Vascular Dysfunction in Preeclampsia. Cells 2021, 10, 3055.
- Nikitina, E.R.; Mikhailov, A.V.; Nikandrova, E.S.; Frolova, E.V.; Fadeev, A.V.; Shman, V.V.; Shilova, V.Y.; Tapilskaya, N.I.; Shapiro, J.I.; Fedorova, O.V.; et al. In preeclampsia endogenous cardiotonic steroids induce vascular fibrosis and impair relaxation of umbilical arteries. J. Hypertens. 2011, 29, 769–776.
- Fedorova, O.V.; Ishkaraeva, V.V.; Grigorova, Y.N.; Reznik, V.A.; Kolodkin, N.I.; Zazerskaya, I.E.; Zernetkina, V.; Agalakova, N.I.; Tapilskaya, N.I.; Adair, C.D.; et al. Antibody to Marinobufagenin Reverses Placenta-Induced Fibrosis of Umbilical Arteries in Preeclampsia. Int. J. Mol. Sci. 2018, 19, 2377.
- Agalakova, N.I.; Grigorova, Y.N.; Ershov, I.A.; Reznik, V.A.; Mikhailova, E.V.; Nadei, O.V.; Samuilovskaya, L.; Romanova, L.A.; Adair, C.D.; Romanova, I.V.; et al. Canrenone Restores Vasorelaxation Impaired by Marinobufagenin in Human Preeclampsia. Int. J. Mol. Sci. 2022, 23, 3336.
- Agalakova, N.I.; Reznik, V.A.; Nadei, O.V.; Ershov, I.A.; Rassokha, O.S.; Vasyutina, M.L.; Ivanov, D.O.; Adair, C.D.; Galagudza, M.M.; Bagrov, A.Y. Antibody against Na/K-ATPase Inhibitor Lowers Blood Pressure and Increases Vascular Fli1 in Experimental Preeclampsia. Am. J. Hypertens. 2020, 33, 514–519.
- Agalakova, N.I.; Reznik, V.A.; Ershov, I.A.; Lupanova, E.A.; Nadei, O.V.; Ivanov, D.O.; Adair, C.D.; Bagrov, A.Y. Silencing of Fli1 Gene Mimics Effects of Preeclampsia and Induces Collagen Synthesis in Human Umbilical Arteries. Am. J. Hypertens. 2022, 35, 828–832.
- Kolmakova, E.V.; Haller, S.T.; Kennedy, D.J.; Isachkina, A.N.; Budny, G.V.; Frolova, E.V.; Nikitina, E.R.; Piecha, G.; Malhotra, D.; Fedorova, O.V.; et al. Endogenous cardiotonic steroids in chronic renal failure. Nephrol. Dial. Transplant. 2011, 26, 2912–2919.
- Fedorova, O.V.; Fadeev, A.V.; Grigorova, Y.N.; Agalakova, N.I.; Konradi, A.O.; Bagrov, A.Y. Marinobufagenin induces vascular fibrosis via a pressure-independent mechanism in NaCl-loaded diabetic rats. J. Cardiovasc. Pharmacol. 2019, 74, 436–442.
- Zhang, Y.; Wei, W.; Shilova, V.; Petrashevskaya, N.N.; Zernetkina, V.I.; Grigorova, Y.N.; Marshall, C.A.; Fenner, R.C.; Lehrmann, E.; Wood, W.H., III; et al. Monoclonal Antibody to Marinobufagenin Downregulates TGFβ Profibrotic Signaling in Left Ventricle and Kidney and Reduces Tissue Remodeling in Salt-Sensitive Hypertension. J. Am. Heart Assoc. 2019, 8, e012138.
- Grigorova, Y.N.; Juhasz, O.; Zernetkina, V.; Fishbein, K.W.; Fedorova, O.V.; Bagrov, A.Y. Monoclonal antibody to an endogenous sodium pump inhibitor marinobufagenin reverses aortic remodeling and stiffness in normotensive rats on a high salt intake. Am. J. Hypertens. 2016, 29, 641–646.