Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Junjie Luo and Version 2 by Lindsay Dong.
High mortality rates due to cardiovascular diseases (CVDs) have attracted worldwide attention. It has been reported that mitochondrial dysfunction is one of the most important mechanisms affecting the pathogenesis of CVDs. Mitochondrial DNA (mtDNA) mutations may result in impaired oxidative phosphorylation (OXPHOS), abnormal respiratory chains, and ATP production. In dysfunctional mitochondria, the electron transport chain (ETC) is uncoupled and the energy supply is reduced, while reactive oxygen species (ROS) production is increased.
- cardiovascular disease
- mitochondrial dysfunction
- mitochondrial DNA mutation
1. Mitochondrial Dysfunctions and CVDs
The pathogenesis of cardiovascular diseases (CVDs) is constantly being explored. Although the pathogenesis of CVDs is not fully understood; a study from the perspective of mitochondrial dysfunction and the analysis of the relationship between mitochondrial dysfunction and CVDs will help to better understand and solve the problems related to CVDs. Here, Figure 1we summarizes the relationships between primary CVDs and mitochondrial dysfunction (Figure 1), and further analyzed the four important features of mitochondria in CVDs in detail.
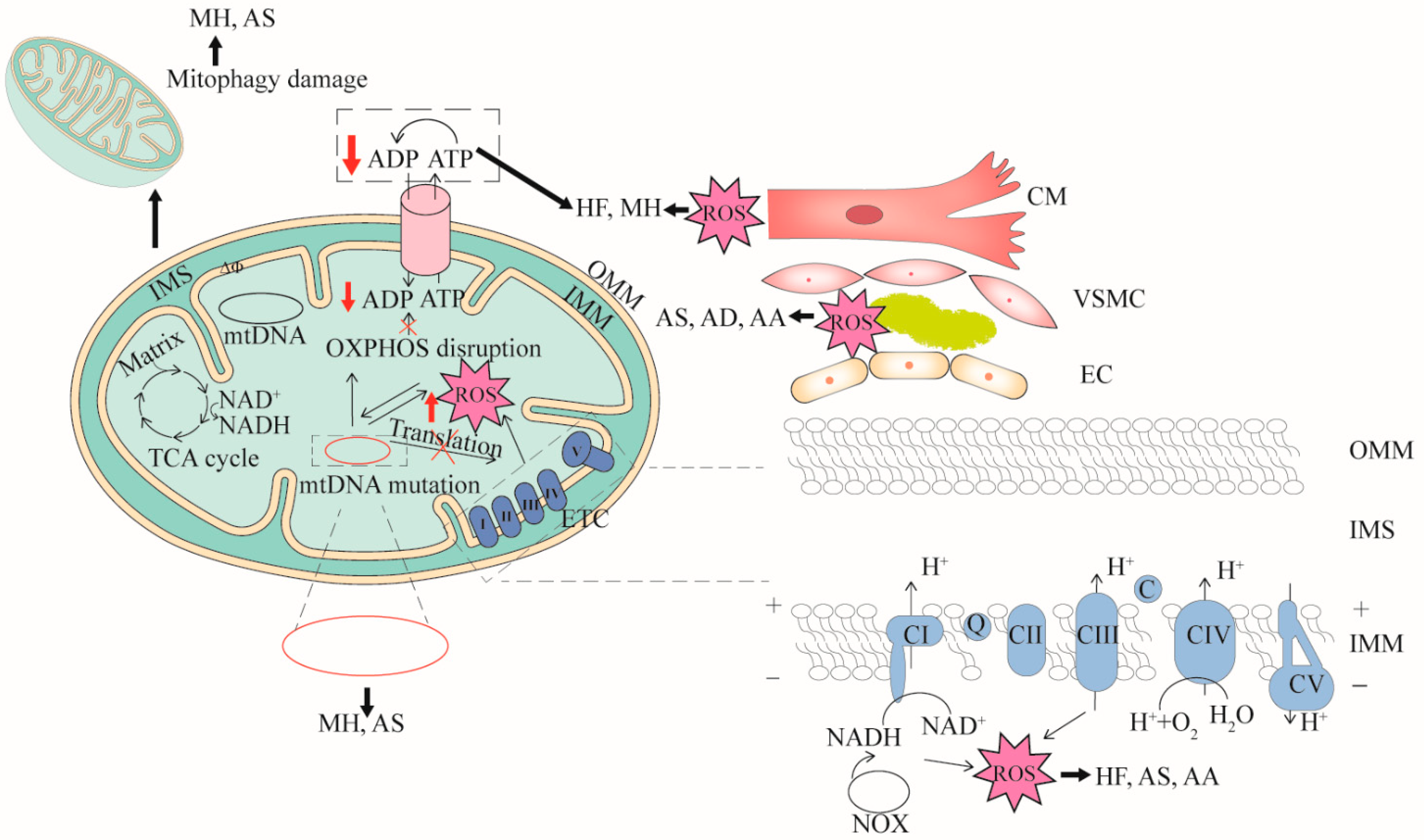
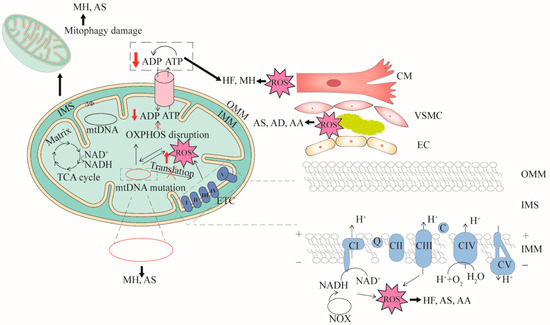
Figure 1. The relationships between mitochondrial dysfunction and CVDs. Four features (mtDNA mutation, mitophagy damage, decreased OXPHOS and increased ROS) associated with mitochondrial dysfunction are demonstrated. mtDNA mutation can cause dysfunction of mitochondrial respiratory chain complex or cytochrome transcription related to OXPHOS. When OXPHOS is impaired, ATP synthesis is reduced and excess ROS is generated. mtDNA mutation directly affects mitochondrial function or ROS production. In turn, high levels of ROS damage mitochondria. Cardiomyocyte cells (CM), vascular smooth muscle cells (VSMC), and endothelial cells (EC) that are impaired by mitochondrial dysfunction can cause CVDs. mtDNA mutation can cause myocardial hypertrophy (MH) and atherosclerosis (AS), mitophagy damage can cause MH and AS, decreased OXPHOS can cause heart failure (HF), aortic aneurysm (AA) and AS, and increased ROS can cause AS, aortic dissection (AD) and AA. OMM, outer mitochondrial membrane; IMS, inter membrane space; IMM, inner mitochondrial membrane.
1.1. mtDNA Mutation in CVDs
Human mtDNA is a circular double stranded genome with a length of 16,569 bp and consists of 37 genes, which support aerobic respiration and the production of cellular energy through OXPHOS. Unlike nuclear DNA, mtDNA is not protected by histones and does not recombine, resulting in approximately 10–100-fold higher mutation rates [1][43]. mtDNA mutations include point mutations deletions, fragment deletion and large scale mtDNA rearrangements, which can directly impair OXPHOS [2][3][44,45]. A large number of mitochondrial diseases are rooted in mtDNA mutations [4][46]. Notably, many specific disease mutations in mtDNA have been observed to cause cardiomyopathy, suggesting that mtDNA encoded proteins play a vital role in mitochondrial function of the heart [5][47]. Heart failure, a complex clinical syndrome that represents the end result of CVDs with multiple etiologies, is a bioenergetic disease with severe mtDNA mutations and mitochondrial dysfunction [6][7][14,48]. The MRPL44-disorder causes problems with the translation of a partial protein participating in OXPHOS, and it is associated with the clinical manifestation of cardiomyopathy in infancy [8][49]. In a mouse model of myocardial infarction (MI), the knockdown of the mouse lncRNA-SNHG8 gene significantly suppressed cardiac tissue injury [9][50]. Myocardial hypertrophy is a common inherited CVDs, and cardiomyocytes are associated with abnormal mitochondrial structure and dysfunction of mitophagy clearance, which makes it impossible to maintain mtDNA and functional integrity [10][4]. Hypertrophic cardiomyopathy is the predominant pattern of cardiomyopathy in mtDNA diseases, observed in nearly 40% of patients [11][51]. The studies have evaluated an association between mtDNA mutations and maternally inherited essential hypertension (MIEH), and these mutations may be one of the pathological mechanisms causing MIEH [3][12][45,52].
1.2. Mitophagy Damage in CVDs
Autophagy plays a positive role in maintaining cellular homeostasis in most cardiovascular-derived cells (e.g., cardiomyocytes, VSMCs) [13][55], and mitophagy is a kind of selective autophagy [14][56]. Mitophagy is one of the mitochondrial quality control pathways, and it can control and remove damaged mitochondria in cells [15][57]. During mitophagy, the damaged mitochondria are sequestered by double membrane vesicles and eventually become hydrolyzed by lysosomes [16][58]. Therefore, if mitophagy is impaired, the accumulation of dysfunctional mitochondria increases, which may lead to abnormal cell function and CVDs. Reducing mitochondrial dysfunction and lipid accumulation by activating mitophagy can help prevent diabetic cardiomyopathy caused by high fat diet [17][59]. In contrast, in BMAL1 deficient hESC-derived cardiomyocytes, impaired mitophagy is a key cause for the development of dilated cardiomyopathy [18][60].1.3. Mitochondrial OXPHOS Reduction in CVDs
Defects in the genes encoding the OXPHOS complex are responsible for triggering various diseases, especially those with high energy requirements [19][63]. Dysfunction of OXPHOS is considered as one of the main causes of CVDs [20][64]. In chronic HF patients, reduced succinyl-CoA levels in myocardial mitochondria cause decreased OXPHOS [21][65].1.4. Mitochondrial-Derived ROS Increase in CVDs
Mitochondrial ROS production is closely related to the mitochondrial ETC and NADPH oxidase (NOX). In the process of mitochondrial electron transfer, complex I and complex III are the main sites of ROS generation [22][23][67,68]. NADPH acts as a substrate to generate ROS under the action of NOX. The NOX is rich in mitochondria, and under the combined action of ETC and NOX, ROS continuously accumulates [24][69]. As a toxic by-product, ROS can damage mitochondria and are involved in the pathomechanism of CVDs. In turn, damaged mitochondria induce a large amount of ROS to be released from adjacent mitochondria, which is known as ROS-induced ROS [25][70]. The increased mitochondrial ROS represents one of the pathogenic mechanisms for vascular diseases [26][71].2. Strategies for Targeting Mitochondria to Treat CVDs
2.1. mtDNA Mutation and Treatment in CVDs
2.1.1. mtDNA Editing Therapy
Mitochondrial heterogeneity affects mtDNA stability through copy number alterations and point deletions [27][91]. Once mtDNA is cleaved and linearized, it is rapidly degraded [28][92]. By duplicating residual mtDNA, mtDNA can be repopulated to the original level. In general, mitochondrial gene editing may include four potential approaches: mitochondria targeted restriction endonuclease (RE) technology, zinc finger nuclease (ZFN) technology, transcription activator-like effector nuclease (TALEN) technology and CRISPR/Cas9 system. In fact, mtDNA editing is a promising therapeutic modality to treat heteroplasmic or mutant mtDNA diseases. Specific mtDNA was effectively eliminated in heart of mice by using a mitochondria targeted RE [29][30][78,93]. Mitochondrial-targeted ZFNs can selectively cleave and degrade pathogenic mtDNA bearing large scale deletions or point mutations [31][94].2.1.2. Mitochondrial Replacement Therapy
mtDNA replacement therapy (MRT) is to use enucleated donor embryos as healthy mtDNA to replace undesired defective/mutated mtDNA to prevent mitochondria from being maternally inherited. MRT is a form of in vitro fertilization (IVF) that includes spindle transfer (ST), prokaryotic transfer (PNT) and polar body transfer (PBT) [32][103]. In fact, embryos from human nuclear transfer can contain low levels of mutated mtDNA, which may be suitable for treating degenerative diseases caused by mtDNA mutations [33][104]. This opens up the possibility of MRT for CVDs, a chronic noncommunicable degenerative disease.2.2. Mitophagy Therapy in CVDs
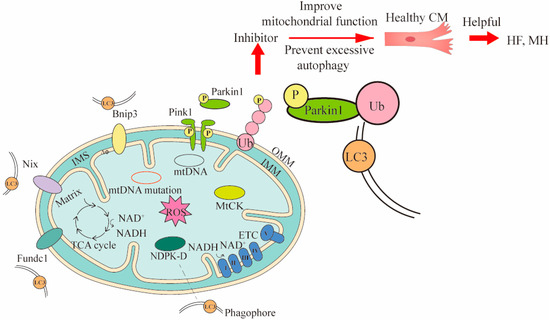
Mitophagy clears dysfunctional mitochondria under normal physiological conditions, and in response to pathological stress [34][15]. Currently, there are three mechanisms of mitophagy, including mitochondrial outer membrane receptor-mediated, Pink1/Parkin pathway, and lipid receptor-mediated mechanisms (Figure 2) [35][109]. Notably, the mechanism mediated by the Pink1/Parkin pathway is the most extensively studied. Under normal circumstances, the content of Pink1 is extremely low. When oxidative stress occurs and mitochondria damage is induced, Pink1 is activated and recruits Parkin to the mitochondrial outer membrane for phosphorylation. The phosphorylated Parkin ubiquitinates the substrate protein on the mitochondrial membrane [36][110]. These ubiquitinated proteins subsequently recruit specific autophagy-related receptors to interact with LC3-II to form autophagosomes [37][111].Figure 2. Three mechanisms of mitophagy and the ways they intervene in treating CVDs. Three mechanisms of mitophagy include mitochondrial outer membrane receptor-mediated (such as Bnip3, Nix and Fundc1), Pink1/Parkin pathway, and lipid receptor-mediated mechanisms (such as MtCK, NDPK-D). The LC3 is located in the phagophore and binds to the corresponding receptor. The LC3 can bind to substances of different mitophagy mechanisms. To demonstrate the mitophagy occurring in CVDs, cardiomyocytes (CM) and Pink1/Parkin pathway were used to intervene in heart failure (HF) and myocardial hypertrophy (MH) by inhibitors. In Pink1/Parkin-dependent mitophagy, the Pink 1 accumulated on the damaged mitochondria is activated and recruits Parkin for phosphorylation. The phosphorylated Parkin binds to the ubiquitin attached to outer OMM, and finally binds to LC3 for mitophagy. The Bnip3 and Nix can directly bind LC3 and promote mitophagy. MtCK and NDPK-D as specific transporters can also directly bind LC3 for mitophagy to eliminate damaged mitochondria. The inhibitor acts on Pink1/Parkin pathway, and then prevents excessive mitophagy and improves mitochondrial function, thereby maintaining the healthy levels of cells associated with CVDs. OMM, outer mitochondrial membrane; IMS, inter membrane space; IMM, inner mitochondrial membrane.
In addition, when abnormal mitochondria undergo fission, they can trigger cardiovascular dysfunction [38][39][113,114]. Cytoplasmic GTPase dynamics related protein 1 (Drp1) regulates mitochondrial fission by interacting with proteins located at fission sites such as mitochondrial fission 1 (Fis1), mitochondrial fission factor (Mff), and mitochondrial dynamics proteins of 49 and 51 kDa (MiD49 and MiD51) [40][115].
2.3. Mitochondrial OXPHOS Reduction and Treatment in CVDs
2.3.1. Small Molecule Compounds Enhance Mitochondrial Function
SIRT3 is a mitochondrial protein deacetylase that regulates mitochondrial function and is considered as an emerging drug target for CVDs [41][118]. SIRT3 can make mitochondrial metabolic pathways and ROS detoxification activate, and increase ATP production [42][119]. Resveratrol improves mitochondrial OXPHOS in diabetic hearts and prevents the decline of SIRT3 activity in the heart by increasing ETC activity and mitochondrial function [43][120]. The polyphenolic compound polydatin can initiate SIRT3-regulated mitophagy to prevent MI [44][84].
When mtDNA is damaged at high levels, increased Poly(ADP-ribose) polymerase (PARP) activity leads to a decrease in NAD+ levels, resulting in impaired NAD+-dependent SIRT3 activation and ultimately cardiac mitochondrial dysfunction [45][121]. Therefore, targeted improvement of mitochondrial function through nutritional supplementation NAD+ or ketoesters may be useful in patients with heart failure [46][87]. NAD+ supplementation with nicotinamide riboside (NR) promotes mitophagy in a Pink1-dependent manner [47][122]. NR can reduce ROS production and maintain normal mitochondrial function in the presence of inflammatory triggers [48][123].