Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Himanshu Arora and Version 2 by Catherine Yang.
Nitric oxide (NO) is a short-lived, ubiquitous signaling molecule that affects numerous critical functions in the body. There are markedly conflicting findings in the literature regarding the bimodal effects of NO in carcinogenesis and tumor progression, which has important consequences for treatment.
- nitric oxide
- prostate cancer
- castration
1. Lung Cancer
Lung cancer has the second-highest incidence amongst all cancer types, with a poor 5-year survival rate of 4–17% [1][2][130,131]. Due to its high incidence and often aggressive course, it is no surprise that NO has been shown to function in the pathogenesis of lung cancer. In fact, population-based studies of NO and NO metabolites suggest that increased accumulation of NO metabolites were associated with an increased risk of lung cancer, even after controlling for relevant confounding factors such as smoking [3][132]. Furthermore, cigarette smoking, a notable risk factor for lung cancer, often contains NO and other ROS [4][133]. Such compounds may encourage the development of NO-mediated cellular changes, which can eventually accumulate in malignant lung tissue (Figure 15) [4][133]. Excess exposure of lung tissue to NO results in the accumulation of nitrosylated proteins, which were significantly higher in those with lung cancer [5][134]. Often, this effect is mediated via iNOS, which is often shown to be upregulated in lung cancer cell lines, similar to other cancers [6][7][125,126].
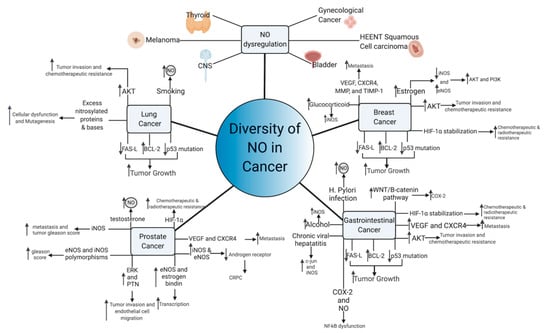
Figure 15. Diversity of NO functioning in cancer. Multiple NO-mediated cancer pathways contribute to cancer growth and metastasis. Common pathways of NO-mediated mutagenesis and cancer growth, include p53 mutation, AKT upregulation, and VEGF induction, among others. Aberrant NO signaling also occurs in response to common carcinogens, including viruses, alcohol, and tobacco, suggesting that NO may lie in a common carcinogenic pathway shared by these compounds. In hormone-sensitive tumors, NO paradoxically functions to transmit the hormonal growth signals and can make the tumor hormone-insensitive.
Excess NO in lung tissues may contribute to a number of effects that synergistically act to encourage cancer formation. For example, one study noted that gaseous nitric oxide and inducible NOS produced an increase of 8-nitroguanine, which may increase DNA damage and encourage mutagenesis [8][135]. Although protein nitrosylation is often viewed as a marker of oxidative stress, one group proposed that protein nitrosylation may also impair antioxidant proteins and those involved in cellular metabolism, which may further contribute to the development of non-small-cell lung cancer (NSCLC) [9][136]. Furthermore, elevated iNOS in NSCLC cells are linked to p53 mutations, removing growth checkpoint inhibition and creating cells with unlimited replicative potential, another notable effect of NO [10][11][137,138]. Within lung cancer, the antiapoptotic effect is hypothesized function through BCL-2 upregulation and modifications to FAS death-ligand signaling, which encourages aggressive growth [12][139]. Once established, NO also modulates integrin expression amongst NSCLC cells via protein kinase B (AKT) activity, and increased AKT activity is correlated with a poor prognosis and chemotherapeutic resistance [13][140]. Increased exposure to nitric oxide over time is itself correlated with enhanced cellular migration and chemotherapy resistance [14][15][141,142]. Nevertheless, a number of NO-donating compounds have been reported to have efficacy at inhibiting some of these effects [16][17][18][143,144,145]. As such, NO-donating drugs may raise the concentration of NO to cytotoxic levels, bypassing the physiological mechanisms underlying tumorigenesis and actually serve to inhibit cancer growth.
2. Breast Cancer
Amongst all non-skin cancer sites in females, breast cancer has the highest incidence and second-highest mortality, although the 5-year survival rate is over 90% [19][129]. Similar to NSCLC and other types of cancer, elevated levels of iNOS were noted from tissue samples of patients with breast cancer, which was significantly greater than normal breast tissue and benign breast diseases such as fibroadenomas [20][123]. NO was also shown to function through the same p53 and AKT pathways as those of lung cancer cells, suggesting common pathways for NO signaling to induce cancer growth and encourage progression [21][146]. Such pathways may work synergistically with other NO pathways in breast cancer, such as the epidermal growth factor receptor (EGFR)-mediated activation of ERK [22][147]. Furthermore, a number of studies suggest that the activity of NO pathways in breast cancer are concentration-dependent, which is backed by other NO studies. NO concentration of less than 100 nM activates cGMP-dependent pathways, while concentrations of 200–600 nM activate cGMP independent pathways [21][146]. Above these concentrations, NO has been suggested to phosphorylate p53 and halt the function of DNA repair enzymes [21][146]. cGMP dependent pathways were noted to be dysregulated within breast cancer, with lower concentration of cGMP being correlated to malignant disease, suggesting low physiological concentration of NO-mediated cGMP signaling may actually provide protection against breast cancer [23][148]. cGMP independent pathways have been discussed previously and are not unique to breast cancer. Nevertheless, NO-mediated HIF-1a stabilization, nitrosylation of metabolic enzymes, and induction of VEGF are major pathways of breast cancer growth and metastasis [21][146]. Additionally, NO-mediated activation of matrix metalloproteinases (MMP) and inhibition of tissue inhibitor of matrix metalloproteinases-1 (TIMP-1) may reorganize the tumor microenvironment to increase metastatic potential, while further stabilizing downstream cGMP-independent NO pathways [21][146]. Numerous studies have found that many of these NO-mediated effectors, such as VEGF and CXCR4, are correlated to lymph node metastasis and overall prognosis [24][25][149,150]. The growth arrest and p53 phosphorylation exhibited by supraphysiological concentrations of NO may explain the effect that NO-conjugated drugs have at inhibiting breast cancer growth and inducing apoptosis [26][151]. The underlying mechanisms behind how nitric oxide becomes upregulated in breast cancer remain unknown; however, similar to other cancer types, systemic effects from environmental, hereditary, and physiological processes all may play a role. For example, iNOS was noted to be upregulated in response to glucocorticoids, a stress hormone, suggesting multisystem processes may contribute to the pathogenesis of breast cancer [27][152]. Furthermore, cyclooxygenase-2 (COX-2) and NO are strongly linked in breast cancer, with one multivariate analysis suggesting that those with COX-2 and iNOS-positive tumors were associated with extremely poor prognosis [28][153]. Nevertheless, the exact role of NO in breast cancer remains controversial, especially in the context of estrogen- and progesterone-mediated signaling.
Estrogen and progesterone have also been shown to modulate nitric oxide expression, through the actions of eNOS and iNOS [29][30][154,155]. Estrogen is thought to downregulate the expression of iNOS and upregulate the expression of eNOS, which in turn activates phosphoinositide 3-kinase (PI3K) and AKT through direct activation and increased transcription, which have been shown to be critical for cancer proliferation [31][156]. Furthermore, upregulation of eNOS appears to be a unique feature of estrogen-dependent tumors, which are lacking in ER- breast cancers [32][157]. Interestingly, progesterone’s function within nitric oxide synthesis appears to be more controversial, with several studies suggesting that progesterone can affect eNOS production and others suggesting null or contradictory effects [31][33][34][156,158,159]. For example, one study suggested that progesterone-induced iNOS expression in vitro, which then induced cell apoptosis [35][160]. Additionally, progesterone has been proposed to affect and downregulate estrogen-mediated nitric oxide production [31][156]. These results suggest that breast cancer tumor progesterone status is a favorable prognostic factor, an effect that is backed by clinical studies [36][161]. Although it is currently unknown exactly how the surface marker human epidermal growth factor receptor 2 (HER2/neu) affects nitric oxide production, one group suggests that HER2/neu downregulates nitric oxide production, ablating the apoptotic effect of chemotherapeutics in vitro, potentially through a COX-2 mechanism [37][38][162,163]. Nevertheless, triple-negative breast cancer, the most aggressive form, appears to also be strongly correlated to iNOS [39][164]. Thus, nitric oxide expression appears to be central to the pathogenesis of breast cancer, regardless of receptor phenotype expression.
3. Prostate Cancer
Prostate cancer is highly prevalent in men and has an age-adjusted incidence of 453.8 per 100,000 and is highest amongst those 65–74 [40][165]. Similar to other cancers, iNOS expression was also significantly increased in prostate adenocarcinoma when compared to healthy prostate tissue [41][166]. Furthermore, previous work demonstrated that iNOS expression was greatest amongst those with metastasis and high Gleason scores, and one meta-analysis found that tumor iNOS expression may serve prognostic value [42][43][167,168]. These findings are backed by clinical studies, which suggest greater nitrosative stress in those with prostate cancer as compared to benign prostatic hyperplasia (BPH) and controls [44][169]. However, eNOS also appears to be linked to prostate cancer, as some but not all studies have shown that genetic polymorphisms of iNOS and eNOS carry an increased risk of high Gleason score prostate cancer [45][46][47][170,171,172]. eNOS may in turn upregulate pleiotrophin (PTN), expression through ERK activity, increasing tumor and endothelial migration, laying the groundwork for metastatic disease [48][173]. Additionally, the eNOS complex can interact with the estrogen receptor to transcriptionally alter other important gene products, such as Glutathione transferase P1, which are correlated to disease pathogenesis [49][174].
Prostate cancer is often initially androgen-sensitive, which is useful in androgen deprivation therapy, and is often a first-line therapy for advanced and metastatic disease [50][175]. This advanced form of prostate cancer is termed castration-resistant (CRPC) and is associated with poor outcomes [51][176]. Previous studies have implicated NO as a potential mediator of androgen resistance by androgen receptor transcriptional suppression and direct androgen receptor inhibition, which were mediated by iNOS and eNOS, respectively [52][53][177,178]. iNOS induction may encourage tumor growth, as two studies from the same lab found that NO promotes survival and accelerates tumor growth after oxidative stress [54][55][179,180]. However, testosterone, an androgen, also increases NO concentration and survival of prostate cancer cells, suggesting that NO’s mechanism in androgen sensitivity and resistance remains elusive [56][57][58][59][1,2,181,182]. These androgen-specific mechanisms may interact with previously defined effects of NO on hypoxia-induced HIF-1a and tumor angiogenesis, compounding the effect of nitric oxide on prostate cancer [60][183].
4. Gastrointestinal Cancers
When summed, digestive system cancers have the highest incidence amongst all organ systems with colorectal cancer having the third-highest incidence amongst all non-skin types, regardless of gender [19][129]. Interestingly, one analysis of gastrointestinal cancers suggested that most had adherent iNOS expression, although whether iNOS expression was upregulated or downregulated was dependent upon cancer type [61][184]. Similar to other cancers, gastrointestinal cancers may also use previously discussed NO signaling pathways such as PI3-K, p53, AKT, PTEN, NF-kB, MMPs, and HIF-1a in their carcinogenic pathogenesis [61][184]. Malignant transformation of colorectal and other cancers are often dependent upon the epithelial–mesenchymal transition (EMT) in which cancer cells express genes that are normally associated with connective tissue [62][185]. Although a number of cell signals can stimulate this pathway, one particularly important player is the APC/Wnt/β-catenin pathway, which is often dysregulated in colorectal cancer [63][186]. In normal healthy colonic tissue, APC downregulates B-catenin, which prevents polyp formation [63][186]. NO has also been shown to upregulate the Wnt/β-catenin pathways, potentially through negative feedback NF-κB response elements on a Dickkopf-1 gene promoter that reduces gene silencing [64][187]. iNOS has been previously suggested to influence the function of NF-κB, and these findings correlate to the increased iNOS expression, which is often noted in colorectal carcinomas. However, increased iNOS expression is not a ubiquitous finding across all studies [65][188]. In addition, APC and the wingless-related integration site (WNT)/B-catenin pathways also serve to regulate COX-2 in a similar manner to NO, suggesting that NO may work in synchrony with COX-2 to promote its pro-cancer effects [61][66][184,189]. COX-2 has been previously linked to many of the same pro-cancer effects as NO [67][190]. Nevertheless, the underlying mechanisms linking COX-2 to NO remain elusive, although both undergo NF-kB regulation [68][191]. Taken together, these findings suggest that regulation of NO and APC may occur simultaneously, and treatment strategies targeting these pathways may prevent WNT/B-catenin upregulation, preventing the development of cancer.
Similar to cigarette smoking, the processing of digested metabolites and chronic infectious agents may also lead to cancer through nitric oxide-mediated mechanisms. For example, one of the major risk factors for gastric cancer is dietary consumption of nitro-compounds, with a relative risk of 1.31 (95% confidence interval (CI), 1.13–1.52) for nitrites, and 1.34 (95% CI, 1.02–1.76) for NDMA, although nitrate consumption was associated with a lower risk of gastric cancer 0.80 (95% CI, 0.69–0.93) [69][192]. Dietary consumption of salivary nitrites exposes nitrites to stomach acid and ascorbic acid, which then produces nitric oxide, which can diffuse rapidly to surrounding tissue [70][193]. A similar increase in NO production was noted with chronic Helicobacte pylori infection, suggesting that enhanced nitric oxide exposure from environmental and infectious agents may be responsible for the development of cancer, especially when there is high or chronic levels of exposure to these agents [71][194]. Viruses were also not immune to NO, as Hepatitis B was shown to be inhibited by NO via INF-γ, and chronic infection with Hepatitis B may induce increased NO, which can predispose hepatocytes to mutagenesis, potentially through a c-jun, an n-terminal protein kinase (JNK) [72][73][195,196]. Furthermore, Hepatitis C was also shown to induce DNA damage through upregulation of iNOS and nitrosylation of DNA glycosylase [73][74][196,197]. The effects of chronic viral-induced upregulation of NO may be compounded by mutations to proteins regulating nitrosylation, like GSNOR, which can cause the buildup of formaldehyde, a well-known carcinogen [73][196], increasing the risk of liver cancer. Evidence also suggests that iNOS and NO may be dysregulated upon alcohol exposure, as iNOS and NO are upregulated in response to ingested toxins [73][75][196,198]. When combined with studies on NO, such results suggest that increased nitric oxide dysregulation in response to cigarettes and alcohol may be one mechanism for the increased cancer risk amongst patients who engage in these behaviors.
5. Other Cancers
NO dysregulation is linked to a wide range of malignancies, including those of the brain [76][199], genitourinary system [77][78][200,201], skin [79][202], thyroid [80][203], and head and neck [81][82][204,205] cancers. Due to the complexity of nitric oxide and its wide number of potential interactions, it is suggested that the multifactorial effects of NO are cancer-specific. Although NO may often work through a common pathway such as p53 to induce mutagenesis, the exact mechanisms often differ between cancer types. Furthermore, in cases of sporadic cancers with minimal risk factors and no underlying conditions, it is unknown exactly what causes the underlying upregulation of NO or if the elevated NO is in response to another effect. However, chronic unchecked NO signaling is clearly beneficial to tumors and can encourage progression and metastasis. Therefore, therapies that can control NO growth signals have great promise, although delivering such therapies to the tumor without affecting nearby or distant healthy cells remains a significant problem.