Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Jessie Wu and Version 4 by Jessie Wu.
Parkinson’s disease (PD) is the second most common neurodegenerative disorder after Alzheimer’s disease. It affects about 1% of the population over the age of 60, with the total number of patients exceeding 6.1 million worldwide. As a highly diverse and complex pathology, PD is represented by a plethora of motor symptoms such as tremor, muscle rigidity, bradykinesia, and postural instability. Non-motor symptoms, including cognitive and behavioral impairments, sleep irregularities, sensory and autonomic dysfunction, are also common in PD.
- Parkinson’s disease (PD)
- neurodegeneration
- neuroinflammation
- YKL-40
1. Introduction
The inflammatory response serves to efficiently eliminate the causative agent and to facilitate tissue repair [1]. The initiation and progression of inflammation depend on the coordinated interaction between immune and non-immune cells and the fine regulation of inflammatory mediators. Primary inflammatory stimuli (molecules and structures of microbial origin, aggregated or misfolded proteins) and cytokines—interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α)—promote inflammation. It is triggered as a result of interaction with the Toll-like receptors (TLRs), IL-1 receptor (IL-1R), IL-6 receptor (IL-6R), and the TNF receptor (TNFR) [2]. The activated receptors activate intracellular signal transduction cascades, including the mitogen-activated protein kinase (MAPK), nuclear factor kappa-B (NF-κB), and Janus kinase signal transducer and activator of transcription (JAK-STAT) pathways. Effector macrophages and lymphocytes release pro- and anti-inflammatory cytokines that recruit other leucocytes and modulate the inflammation itself via a complex network of interactions, thus regulating both the expansion and the intensity of the process [3].
The inflammatory response in the central nervous system (CNS) (neuroinflammation) has been directly associated with viral and bacterial diseases, autoimmune and neurodegenerative conditions, trauma, vascular damage, and neuropsychiatric disorders. Neuroinflammation can in-crease the neuronal excitability, trigger cellular damages, and augment the permeability of the blood-brain barrier [4].
Different studies have demonstrated that neuroinflammation participates not only in typically inflammatory diseases such as viral encephalitis but also in neurodegenerative conditions, including Parkinson’s disease (PD) [5][6]. Neuroinflammation in PD involves activation of microglia and T-lymphocytes alongside an increased expression of pro-inflammatory cytokines. Experiments with animal models of PD have indicated that neuroinflammation is profoundly involved in neuronal cell death, despite not being its primary cause. In agreement with this assumption, available evidence suggests a significant role of glucocorticoid receptors in modulating microglial reactivity and their substantial dysregulation in the inflammation-mediated neuronal degeneration [7].
2. Microglia in Parkinson’s Disease
Microglial cells are the resident macrophages of the brain [8]. First discovered by Pío del Río Hortega [9], they serve as primary cells of innate immunity in the CNS and play a crucial role in maintaining the homeostasis of the brain [10]. Microglia participate in synaptogenesis, synaptic pruning, neural progenitor-cell growth and differentiation, and myelinogenesis [11][12][13]. Microglial activation is a complex response against infection or injury that produces two functionally distinct phenotypes: M1 and M2 [14]. According to the general model, although greatly simplified, M1 microglia secrete pro-inflammatory cytokines (IL-1β, IL-6, IL-12, TNFα) that stimulate neurodegeneration [15]. These mediators broaden the immune response and may directly contribute to neuronal death. TNFα is known for its pro-apoptotic activity which, in neurons, depends on the downregulation of c-Rel, a NF-κB homologue that inhibits cell death and promotes neuronal survival [16]. M1 cells also upregulate enzymes that produce reactive oxygen species with antimicrobial function, thus elevating oxidative stress. Simultaneously, microglial metabolism shifts from oxidative phosphorylation (OXPHOS) to glycolysis, allowing microglia to adapt to increased energy demands. Metabolic reprogramming leads to faster, although less efficient, production of ATP for proliferation, cytokine production, and ROS generation [17][18]. It has been demonstrated that the glycolytic inhibitor deoxy-D-glucose (2-DG) decreases TNFα and IL-6 in microglia through NF-κB suppression, inducing microglial death [19]. Furthermore, an in vitro study on BV-2 microglial cells revealed an elevated lactate production and decreased mitochondrial activity following lipopolysaccharide stimulation [20]. In contrast with M1, M2 cells express factors involved in the inhibition of inflammation and promotion of tissue repair. They secrete substances, such as IL-10, to reduce the activity of pro-inflammatory cells. M2 microglia also express high levels of phagocytic receptors to promote the clearance of cell debris [21]. Nevertheless, high-throughput studies have revealed that microglial heterogeneity is even more complex, suggesting the presence of a wider spectrum of microglial phenotypes [22]. To date, little is known about the molecular mechanisms of microglial heterogeneity.
Studies of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD mice models have demonstrated that microglial activation is a prominent and persistent feature of PD [23][24][25]. Even the fact that substantia nigra constitutes the predominantly affected site in PD is in concordance with the higher abundance of microglial cells in this brain region [26]. Microglia exert versatile roles in neuroinflammation, serving as both a damaging and a protective factor. When activated, microglial cells infiltrate the site of neuroinflammation, where they perform phagocytosis and secrete both pro- and anti-inflammatory cytokines [27]. The cytokine synthesis and secretion are hallmarks of microglial activation as part of the early inflammatory response and persist throughout the disease progression [28][29][30]. Secretion of pro-inflammatory factors such as IL-1β, IL-12, TNFα, and inducible nitric oxide synthase (iNOS) greatly stimulates neuroinflammation and often corresponds to significant neuronal loss. Conversely, the production of anti-inflammatory cytokines such as IL-4, IL-10, IL-13, TGFβ, and IGF-1 by microglial cells suppresses inflammation and promotes neuroprotection [27]. From this perspective, a prominent factor that may contribute to microglial activation is the release of α-Syn. It is an abundant neuronal protein that localizes to the presynaptic terminals in the CNS where it regulates vesicular release [31][32][33]. Its native conformation is largely unfolded but the same protein can also exist in abnormal aggregated forms such as oligomers, protofibrils, and fibrils [34]. α-Syn is the main component of Lewy bodies and, as such, significantly contributes to the pathogenesis of PD. In the PD brain, α-Syn is often released by neurons in the extracellular interstitium, which allows its laboratory detection in the bodily fluids of PD patients. Subsequently, the α-Syn-induced microglial activation triggers rapid α-Syn phagocytosis. In this process, the activated microglial cells engage their FcγR receptors in the α-Syn uptake and initiate a sequence of pro-inflammatory events such as NF-κB/p65 translocation and increased secretion of cytokines. These neuroinflammatory effects then result in neuronal loss and chronic neurodegeneration in PD. In addition to α-Syn, other PD risk factors such as DJ-1 and LRRK2 can also participate in the regulation of microglia-mediated inflammation. For instance, LRRK2 deficiency represses inflammation by inhibiting the p38 MAPK and NF-κB pathways [27].
3. Astroglia in Parkinson’s Disease
Astroglia constitute the largest population of glial cells in the brain and perform functions essential for the normal physiology of the CNS. Astrocytes mechanically support the neurons and the adjacent capillaries. They maintain the integrity of the blood–brain barrier and its permeability [10][35][36]. Astroglia synthesize and secrete a plethora of neurotrophic factors, including glial cell line-derived neurotrophic factor (GDNF), brain-derived neurotrophic factor (BDNF), nerve growth factor (NGF), and cerebral dopamine neurotrophic factor (CDNF). These neurogenic molecules stimulate and fine tune neuronal development, survival, and plasticity. In addition, CDNF provides neuroprotection and promotes the recovery of damaged dopaminergic neurons [37][38][39][40]. In the structure of the tripartite synapse, astrocytes surround the synaptic cleft where they interact with the pre-and post-synaptic neurons and uptake excessive glutamate [36]. Astrocytes also provide metabolic support for the neurons by transferring lactate for the Krebs cycle. They are able to produce antioxidants and to neutralize neuronal waste products, including aggregated α-Syn and damaged mitochondria [41][42]. Finally, astrocytes are responsible for the remodeling of the nervous tissue by filling the gaps left after neuronal death, forming the so-called astroglial scar [43].
Similar to microglia, astrocytes exist in different functional states. The A1 astrocytic population produces pro-inflammatory factors such as IL-1α, C1q, and TNFα, thus enhancing neuronal death and inflammation. Conversely, the A2 population promotes neuronal survival and neuroprotection after injury [44]. Liddelow et al. (2017) have determined that microglia cause astrocytic activation by secreting the cytokines IL-1α, TNF, and C1q [43]. The authors also demonstrated an elevated production of pro-inflammatory cytokines such as TNF-α, IL-1α, and IL-1β in A1 astrocytes as a consequence of this activation. In their pro-inflammatory state, astrocytes no longer assist neuronal survival but induce cell death by releasing neurotoxic molecules. In turn, astrocytes can modulate microglial activation and microglia-mediated inflammation [44].
Altered astrocytic function is involved in different mechanisms of PD development, such as α-Syn accumulation, neuroinflammation, impaired mitochondrial metabolism and oxidative stress. Of particular interest is the fact that at least eight out of 17 genes of known causative importance for PD are expressed in astrocytes [45]. One of them, PARK7, is even more prominent in astroglia than in neurons and shows noticeable upregulation in astrocytes from PD individuals. The product of this gene, DJ-1, is involved in oxidative stress response, glutamate uptake, and neuroprotection [46].
It has been shown that microglia and astrocytes can exhibit a protective effect on neurons by eliminating extracellular α-Syn. Glial cells engulf and degrade complexes of aggregated α-Syn via proteasomal and autophagic mechanisms [41][42]. Exchange of intracellular materials including α-Syn and intact mitochondria occurs not only between astrocytes and neurons but between neurons themselves [47]. Although α-Syn is predominantly expressed and accumulated in neurons, different studies have reported that α-synuclein aggregates in astrocytes as well. Accumulation of α-Syn can disrupt astrocyte function and accelerate neurodegeneration through mitochondrial dysfunction and impaired autophagy [48].
Sonninen et al. (2020) have demonstrated that metabolic changes occur in iPSC-derived astrocytes from PD patients carrying mutant variants of the LRRK2 gene. These astrocytes were characterized by abnormal α-Syn expression, metabolic alterations, impaired Ca2+ regulation, and elevated cytokine production [49]. It has been proposed that mitochondrial dysfunction in astrocytes possibly evokes neuronal toxicity by altering the normal glutamate uptake and degradation, Ca2+-induced cell death, impaired metabolism, and accumulation of ROS and toxic fatty acids [50]. The interplay between astroglia and microglia is presented in Figure 1.
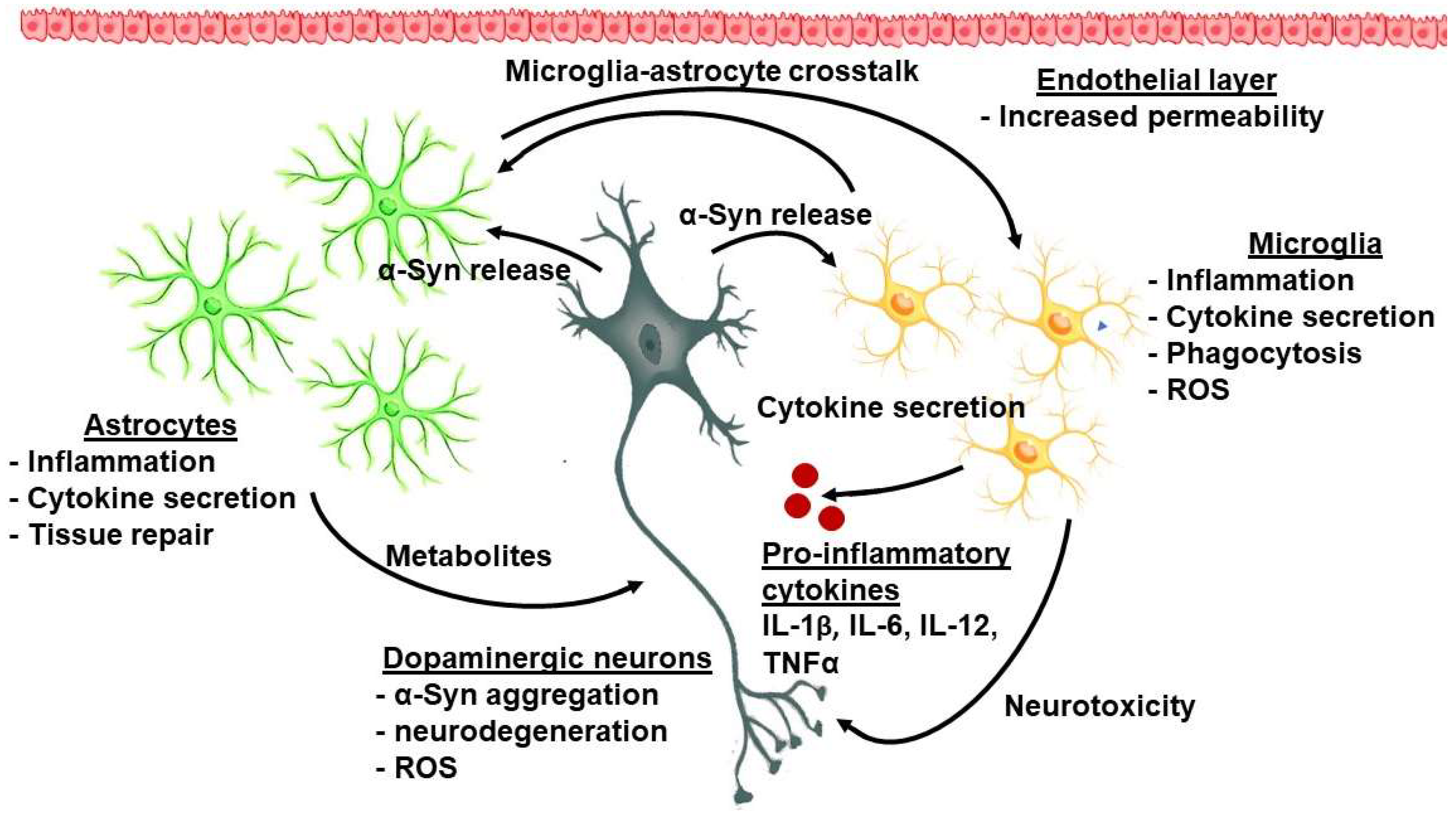
Figure 1. The complex interplay between cellular and molecular factors in PD-related neuroinflammation.
4. Inflammatory Cytokines
Numerous reports have revealed the significant association between PD severity and the level of immune markers in plasma and serum.
Notably higher serum levels of the proinflammatory cytokine IL-1β are detected in patients with early PD [51]. However, no significant correlation was found between the IL-1β levels and the clinical scales for PD assessment. Selikhova et al. (2002) described elevated IL-6 in the plasma of patients with early idiopathic PD [52]. Higher abundance of the pro-inflammatory cytokines IL-1β, INF-γ, and TNF-α was detected in PBMCs isolated from PD patients [53]. The levels of TNF-α (but not those of IL-1β and IL-10) correlate with cognition and other non-motor symptoms of PD [54].
It is noteworthy that, contrary to these findings, a significant downregulation of inflammatory cytokines has also been demonstrated in patients with PD. In a study by Rocha et al. (2018), the PD-patient cohort exhibited a lower percentage of T-lymphocytes, including activated T-lymphocytes, in comparison with healthy controls [55]. In accordance with these findings, the authors also described decreased plasma levels of IL-4, IL-6, IL-10, TNF, IFN-γ, and IL-17A in the PD group [55]. In a previous paper, the same authors also detected significantly elevated levels of the soluble TNF-α receptors, sTNFR1 and sTNFR2, in plasma of PD patients, suggesting the inflammatory etiology of PD [56]. Another comparative analysis [57] suggested that IL-6 was significantly higher in patients with PD than in healthy controls. Conversely, the authors found no significant differences in the levels of CRP, sIL-2R, or TNF-α between the two studied groups. Dufek et al. (2008) investigated a panel of inflammatory markers in serum samples from 29 patients with PD and found significant overexpression of only TNF-α [58]. None of the other markers of interest (IL-6, acute phase proteins, and factors of the complement system) showed any abnormal changes in the PD group. There were also no significant correlations between the patients’ clinical state and the levels of the examined serum markers.
Another cohort study revealed significantly lower serum levels of IL-1α and IL-6 in PD patients than in their age-matched controls [59]. Conversely, the serum IL-1β levels in the PD group appeared significantly higher than those in the control one. Again, the authors observed no correlation between the studied markers and disease severity.
In a study of 83 PD patients and 83 healthy subjects, higher serum levels of TNF-α and lower levels of IL-27 were detected in patients with PD compared to healthy controls (p < 0.0001) [60].
All these studies are greatly limited due to the relatively small number of participants. The first large-cohort research to evaluate serum cytokine markers in the context of PD examined 262 newly diagnosed PD patients and 99 healthy controls [61]. It demonstrated that a panel of cytokines is robustly associated with cognitive and motor features of PD. The experimental results revealed higher levels of TNF-α, IL-1β, IL-2, and IL-10 in PD versus healthy individuals. Based on their data, the authors suggest that a more pro-inflammatory profile is associated with impaired cognition and rapid motor regression, while a more anti-inflammatory profile is related to improved cognitive abilities and preserved motor function. Earlier research investigating the role of CRP in 375 PD individuals has suggested that CRP associates with faster motor decline.
The immense scientific work in clarifying the complex interplay between immunity and PD development delineates the potential usefulness of cytokines as biomarkers of inflammation and neurodegeneration. Probably the most promising future insights will combine clinical data, cellular and molecular features.
5. YKL-40 in Parkinson’s Disease-Related Neuroinflammation
The YKL-40 glycoprotein has been established as a prospective biomarker of neuroinflammation in neurodegenerative diseases. YKL-40 has also been debated as a biomarker in diverse medical conditions, including toxoplasmosis [62], autoimmune disorders [63] and hemodialysis inflammation [64]. This protein serves as an acute-phase factor, secreted by a variety of immune cells (especially macrophages) in response to pro-inflammatory signals including IL-1β, IL-6 and IFNγ, and TNFα. It is noteworthy to mention that YKL-40 has a number of different cellular sources (chondrocytes, fibroblast-like synovial cells, vascular smooth muscle cells, and macrophages) [65][66]. The previous results showed correlation of YKL-40 and neuron-specific enolase levels with clinical scores for assessment of severity and outcome of traumatic brain injury [67]. Researchers proposed that YKL-40 might reflect certain aspects of the response to brain injury, such as neuroinflammation and brain damage. A series of studies have suggested that the levels of YKL-40 correlate with the glial activation and the number of cells involved in neurodegeneration [68][69][70]. Its levels in cerebrospinal fluid (CSF) have been correlated with the disease phenotype of Parkinson’s-related disorders. For instance, Magdalinu et al. (2015) discovered that the levels of YKL-40 were lower in patients with PD compared to those with atypical Parkinson’s syndrome, but still higher than the levels in the control group [68]. No correlations with disease stages or severity were observed in this research. However, the expression data regarding YKL-40 in PD remain controversial. Substantially higher YKL-40 levels in PD patients have also been reported. A two-year follow-up study revealed a significant increase in the concentration of YKL-40 in the CSF of PD patients compared to the baseline levels. Furthermore, the steady increase in YKL-40 levels correlated with the deterioration of cognitive abilities [69]. Conversely, according to other authors, the levels of YKL-40 were lower in patients with PD than in healthy controls or those with multisystem atrophy, progressive supranuclear palsy and corticobasal degeneration. Additionally, the concentration of YKL-40 in CSF appeared lower in degenerative disorders known as synucleinopathies than in tauopathies [70]. In that study, Olsson et al. (2013) evaluated the levels of YKL-40 together with those of the soluble CD14 as markers for astrocyte and microglial activation. They examined CNF and serum samples from 37 controls, 50 PD patients, and 79 P+ patients (with progressive supranuclear palsy, corticobasal degeneration, and multiple system atrophy). The experimental results identified significantly lower YKL-40 levels in the CNF of PD patients compared to healthy controls or participants with multiple system atrophy and tauopathies. A more recent study has reported elevated YKL-40 levels in cerebrospinal fluid (CSF) from patients with AD dementia, but not in those with PD and Lewy body dementia (LBD), in comparison with non-dementia controls. The authors also investigated the possible association between YKL-40 dysregulation in CSF and other inflammatory-markers. They found no relationship between YKL-40 and levels of the astrocytic marker glial-fibrillary acidic protein (GFAP), interleukin-8 (IL-8), monocyte chemoattractant protein-1 (MCP-1), and interferon gamma-induced protein 10 (IP-10) [71]. Additionally, the plasma levels of YKL-40 have been evaluated in the extended spectrum of neurodegenerative dementias. Villar-Piqué et al. (2019) described significantly higher plasma YKL-40 levels in Creutzfeldt-Jakob disease (CJD) with a moderate potential to discriminate CJD cases from controls. Additionally, YKL-40 levels were strongly associated with age but not with gender. In CJD, YKL-40 concentrations appear significantly higher at late disease stages [72].
Based on these vast experimental data, the protein YKL-40 may have a potential role as a promising biomarker that reflects the severity of inflammation in PD.
References
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017, 9, 7204–7218.
- Kaminska, B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy—From molecular mechanisms to therapeutic benefits. Biochim. Biophys. Acta 2005, 1754, 253–262.
- Henríquez-Olguín, C.; Altamirano, F.; Valladares, D.; López, J.R.; Allen, P.D.; Jaimovich, E. Altered ROS production, NF-κB activation and interleukin-6 gene expression induced by electrical stimulation in dystrophic mdx skeletal muscle cells. BBA-Mol. Basis Disc. 2015, 1852, 1410–1419.
- Kim, S.Y.; Buckwalter, M.; Soreq, H.; Vezzani, A.; Kaufer, D. Blood-brain barrier dysfunction-induced inflammatory signaling in brain pathology and epileptogenesis. Epilepsia 2012, 53 (Suppl. S6), 37–44.
- Pinteac, R.; Montalban, X.; Comabella, M. Chitinases and chitinase-like proteins as biomarkers in neurologic disorders. Neurol.-Neuroimmunol. Neuroinflamm. 2020, 8, e921.
- Chhatbar, C.; Prinz, M. The roles of microglia in viral encephalitis: From sensome to therapeutic targeting. Cell. Mol. Immunol. 2021, 18, 250–258.
- Hirsch, E.C.; Vyas, S.; Hunot, S. Neuroinflammation in Parkinson’s disease. Park. Relat. Disord. 2012, 18, S210–S212.
- Troncoso-Escudero, P.; Parra, A.; Nassif, M.; Vidal, R.L. Outside in: Unraveling the Role of Neuroinflammation in the Progression of Parkinson’s Disease. Front. Neurol. 2018, 9, 860.
- Bereciartu, J.D.R.-H. Pío del Río-Hortega: The Revolution of Glia. Anat. Rec. 2020, 303, 1232–1241.
- Pajares, M.; Rojo, A.I.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687.
- Ullian, E.M.; Christopherson, K.S.; Barres, B.A. Role for glia in synaptogenesis. Glia 2004, 47, 209–216.
- Le, W.; Wu, J.; Tang, Y. Protective Microglia and Their Regulation in Parkinson’s Disease. Front. Mol. Neurosci. 2016, 9, 89.
- Chounchay, S.; Noctor, S.C.; Chutabhakdikul, N. Microglia enhances proliferation of neural progenitor cells in an in vitro model of hypoxic-ischemic injury. EXCLI J. 2020, 19, 950–961.
- Tang, Y. Editorial: Microglial Polarization in the Pathogenesis and Therapeutics of Neurodegenerative Diseases. Front. Aging Neurosci. 2018, 10, 154.
- Wang, W.-Y.; Tan, M.-S.; Yu, J.-T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136.
- Shih, R.-H.; Wang, C.-Y.; Yang, C.-M. NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front. Mol. Neurosci. 2015, 8, 77.
- Lauro, C.; Limatola, C. Metabolic Reprograming of Microglia in the Regulation of the Innate Inflammatory Response. Front. Immunol. 2020, 11, 493.
- Vilalta, A.; Brown, G.C. Deoxyglucose prevents neurodegeneration in culture by eliminating microglia. J. Neuroinflamm. 2014, 11, 58.
- Wang, Q.; Zhao, Y.; Sun, M.; Liu, S.; Li, B.; Zhang, L.; Yang, L. 2-Deoxy-d-glucose attenuates sevoflurane-induced neuroinflammation through nuclear factor-kappa B pathway in vitro. Toxicol. Vitr. 2014, 28, 1183–1189.
- Voloboueva, L.A.; Emery, J.F.; Sun, X.; Giffard, R.G. Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mitochondrial glucose-regulated protein 75/mortalin. FEBS Lett. 2013, 587, 756–762.
- Akhmetzyanova, E.; Kletenkov, K.; Mukhamedshina, Y.; Rizvanov, A. Different Approaches to Modulation of Microglia Phenotypes after Spinal Cord Injury. Front. Syst. Neurosci. 2019, 13, 37.
- Dubbelaar, M.; Kracht, L.; Eggen, B.J.L.; Boddeke, E.W.G.M. The Kaleidoscope of Microglial Phenotypes. Front. Immunol. 2018, 9, 1753.
- Członkowska, A.; Kohutnicka, M.; Kurkowska-Jastrzebska, I.; Członkowski, A. Microglial reaction in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced Parkinson’s disease mice model. Neurodegeneration 1996, 5, 137–143.
- Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Tieu, K.; Teismann, P.; Vadseth, C.; Choi, D.-K.; Ischiropoulos, H.; Przedborski, S. Blockade of Microglial Activation Is Neuroprotective in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model of Parkinson Disease. J. Neurosci. 2002, 22, 1763–1771.
- Liu, W.; Wei, S.; Huang, G.; Liu, L.; Gu, C.; Shen, Y.; Wang, X.; Xia, S.; Xie, A.; Hu, L.; et al. BMAL1 regulation of microglia-mediated neuroinflammation in MPTP-induced Parkinson’s disease mouse model. FASEB J. 2020, 34, 6570–6581.
- Wang, Q.; Oyarzabal, E.; Wilson, B.; Qian, L.; Hong, J.-S. Substance P enhances microglial density in the substantia nigra through neurokinin-1 receptor/NADPH oxidase-mediated chemotaxis in mice. Clin. Sci. 2015, 129, 757–767.
- Ho, M. Microglia in Parkinson’s Disease. In Neuroglia in Neurodegenerative Diseases, 1st ed.; Advances in Experimental Medicine and Biology; Verkhratsky, A., Ho, M.S., Zorec, R., Parpura, V., Eds.; Springer Nature Singapore: Singapore, 2019; pp. 335–353.
- Hunot, S.; Boissière, F.; Faucheux, B.; Brugg, B.; Mouatt-Prigent, A.; Agid, Y.; Hirsch, E. Nitric oxide synthase and neuronal vulnerability in parkinson’s disease. Neuroscience 1996, 72, 355–363.
- Knott, C.; Stern, G.; Wilkin, G.P. Inflammatory regulators in Parkinson’s disease: iNOS.; lipocortin-1.; and cyclooxygenases-1 and -2. Mol. Cell Neurosci. 2000, 16, 724–739.
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003, 106, 518–526.
- Clayton, D.F.; George, J.M. The synucleins: A family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998, 21, 249–254.
- Clayton, D.F.; George, J.M. Synucleins in synaptic plasticity and neurodegenerative disorders. J. Neurosci. Res. 1999, 58, 120–129.
- Snead, D.; Eliezer, D. Alpha-synuclein function and dysfunction on cellular membranes. Exp. Neurobiol. 2014, 23, 292–313.
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.; Follenzi, A.; et al. Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. J. Clin. Investig. 2008, 118, 777–788.
- Bolaños, J.P. Bioenergetics and redox adaptations of astrocytes to neuronal activity. J. Neurochem. 2016, 139 (Suppl. S2), 115–125.
- Miyazaki, I.; Asanuma, M. Neuron-Astrocyte Interactions in Parkinson’s Disease. Cells 2020, 9, 2623.
- Zhang, G.-L.; Wang, L.-H.; Liu, X.-Y.; Zhang, Y.-X.; Hu, M.-Y.; Liu, L.; Fang, Y.-Y.; Mu, Y.; Zhao, Y.; Huang, S.-H.; et al. Cerebral Dopamine Neurotrophic Factor (CDNF) Has Neuroprotective Effects against Cerebral Ischemia that May Occur through the Endoplasmic Reticulum Stress Pathway. Int. J. Mol. Sci. 2018, 19, 1905.
- Petrova, P.S.; Raibekas, A.; Pevsner, J.; Vigo, N.; Anafi, M.; Moore, M.K.; Peaire, A.E.; Shridhar, V.; Smith, D.I.; Kelly, J.; et al. MANF: A New Mesencephalic, Astrocyte-Derived Neurotrophic Factor with Selectivity for Dopaminergic Neurons. J. Mol. Neurosci. 2003, 20, 173–188.
- Lindholm, P.; Voutilainen, M.H.; Laurén, J.; Peränen, J.; Leppänen, V.-M.; Andressoo, J.-O.; Lindahl, M.; Janhunen, S.; Kalkkinen, N.; Timmusk, T.; et al. Novel neurotrophic factor CDNF protects and rescues midbrain dopamine neurons in vivo. Nature 2007, 448, 73–77.
- Voutilainen, M.; Bäck, S.; Pörsti, E.; Toppinen, L.; Lindgren, L.; Lindholm, P.; Peränen, J.; Saarma, M.; Tuominen, R.K. Mesencephalic Astrocyte-Derived Neurotrophic Factor Is Neurorestorative in Rat Model of Parkinson’s Disease. J. Neurosci. 2009, 29, 9651–9659.
- Morales, I.; Sanchez, A.; Puertas-Avendaño, R.; Rodriguez-Sabate, C.; Perez-Barreto, A.; Rodriguez, M. Neuroglial transmitophagy and Parkinson’s disease. Glia 2020, 68, 2277–2299.
- Hua, J.; Yin, N.; Xu, S.; Chen, Q.; Tao, T.; Zhang, J.; Ding, J.; Fan, Y.; Hu, G. Enhancing the Astrocytic Clearance of Extracellular α-Synuclein Aggregates by Ginkgolides Attenuates Neural Cell Injury. Cell. Mol. Neurobiol. 2019, 39, 1017–1028.
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016, 532, 195–200.
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487.
- Booth, H.D.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370.
- Bandopadhyay, R.; Kingsbury, A.E.; Cookson, M.R.; Reid, A.R.; Evans, I.M.; Hope, A.D.; Pittman, A.M.; Lashley, T.; Canet-Aviles, R.; Miller, D.W.; et al. The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain 2004, 127, 420–430.
- Lee, H.-J.; Suk, J.-E.; Patrick, C.; Bae, E.-J.; Cho, J.-H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.-J. Direct Transfer of α-Synuclein from Neuron to Astroglia Causes Inflammatory Responses in Synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272.
- Wang, C.; Yang, T.; Liang, M.; Xie, J.; Song, N. Astrocyte dysfunction in Parkinson’s disease: From the perspectives of transmitted α-synuclein and genetic modulation. Transl. Neurodegener. 2021, 10, 39.
- Sonninen, T.-M.; Hämäläinen, R.H.; Koskuvi, M.; Oksanen, M.; Shakirzyanova, A.; Wojciechowski, S.; Puttonen, K.; Naumenko, N.; Goldsteins, G.; Laham-Karam, N.; et al. Metabolic alterations in Parkinson’s disease astrocytes. Sci. Rep. 2020, 10, 14474.
- Bantle, C.M.; Hirst, W.D.; Weihofen, A.; Shlevkov, E. Mitochondrial Dysfunction in Astrocytes: A Role in Parkinson’s Disease? Front. Cell Dev. Biol. 2021, 8, 608026.
- Kim, R.; Kim, H.-J.; Kim, A.; Jang, M.; Kim, A.; Kim, Y.; Yoo, D.; Im, J.H.; Choi, J.-H.; Jeon, B. Peripheral blood inflammatory markers in early Parkinson’s disease. J. Clin. Neurosci. 2018, 58, 30–33.
- Selikhova, M.V.; Kushlinskii, N.; Lyubimova, N.V.; Gusev, E.I. Impaired production of plasma interleukin-6 in patients with Parkinson’s disease. Bull. Exp. Biol. Med. 2002, 133, 81–83.
- Reale, M.; Iarlori, C.; Thomas, A.; Gambi, D.; Perfetti, B.; Di Nicola, M.; Onofrj, M. Peripheral cytokines profile in Parkinson’s disease. Brain Behav. Immun. 2009, 23, 55–63.
- Menza, M.; Dobkin, R.; Marin, H.; Mark, M.H.; Gara, M.; Bienfait, K.; Dicke, A.; Kusnekov, A. The Role of Inflammatory Cytokines in Cognition and other Non-Motor Symptoms of Parkinson’s Disease. J. Psychosom. Res. 2010, 51, 474–479.
- Rocha, N.P.; Assis, F.; Scalzo, P.L.; Vieira, L.M.; Barbosa, I.G.; de Souza, M.S.; Christo, P.P.; Reis, H.J.; Teixeira, A.L. Reduced Activated T Lymphocytes (CD4+CD25+) and Plasma Levels of Cytokines in Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 1488–1497.
- Rocha, N.P.; Teixeira, A.L.; Scalzo, P.L.; Barbosa, I.G.; de Sousa, M.S.; Morato, I.B.; Vieira, L.M.; Christo, P.P.; Palotás, A.; Reis, H.J. Plasma levels of soluble tumor necrosis factor receptors are associated with cognitive performance in Parkinson’s disease. Mov. Disord. 2014, 29, 527–531.
- Lindqvist, D.; Kaufman, E.; Brundin, L.; Hall, S.; Surova, Y.; Hansson, O. Non-Motor Symptoms in Patients with Parkinson’s Disease—Correlations with Inflammatory Cytokines in Serum. PLoS ONE 2012, 7, e47387.
- Dufek, M.; Hamanová, M.; Lokaj, J.; Goldemund, D.; Rektorová, I.; Michálková, Z.; Sheardová, K.; Rektor, I. Serum inflammatory biomarkers in Parkinson’s disease. Park. Relat. Disord. 2009, 15, 318–320.
- Dursun, E.; Gezen-Ak, D.; Hanağası, H.; Bilgiç, B.; Lohmann, E.; Ertan, S.; Atasoy, I.L.; Alaylıoğlu, M.; Araz, S.; Önal, B.; et al. The interleukin 1 alpha, interleukin 1 beta, interleukin 6 and alpha-2-macroglobulin serum levels in patients with early or late onset Alzheimer’s disease, mild cognitive impairment or Parkinson’s disease. J. Neuroimmunol. 2015, 283, 50–57.
- Kouchaki, E.; Kakhaki, R.D.; Tamtaji, O.R.; Dadgostar, E.; Behnam, M.; Nikoueinejad, H.; Akbari, H. Increased serum levels of TNF-α and decreased serum levels of IL-27 in patients with Parkinson disease and their correlation with disease severity. Clin. Neurol. Neurosurg. 2018, 166, 76–79.
- Williams-Gray, C.H.; Wijeyekoon, R.; Yarnall, A.J.; Lawson, R.A.; Breen, D.P.; Evans, J.R.; Cummins, G.A.; Duncan, G.W.; Khoo, T.K.; Burn, D.J.; et al. Serum immune markers and disease progression in an incident Parkinson’s disease cohort (ICICLE-PD). Mov. Disord. 2016, 31, 995–1003.
- YKL-40 as a novel diagnostic biomarker in Toxoplasmosis. J. Popul. Ther. Clin. Pharmacol. 2022, 29, e61–e70.
- Tizaoui, K.; Yang, J.W.; Lee, K.H.; Kim, J.H.; Kim, M.; Yoon, S.; Jung, Y.; Park, J.B.; An, K.; Choi, H.; et al. The role of YKL-40 in the pathogenesis of autoimmune diseases: A comprehensive review. Int. J. Biol. Sci. 2022, 18, 3731–3746.
- Yamada, K.; Hyodo, T.; Urabe, S.; Haga, S.; Hosaka, T. Serum YKL-40 Level is Associated with Geriatric Nutritional Risk Index (GNRI) and γ-GTP in Hemodialysis Patients. J. Med. Investig. 2022, 69, 101–106.
- Kazakova, M.H.; Sarafian, V.S. YKL-40—A novel biomarker in clinical practice? Folia Med. 2009, 51, 5–14.
- Väänänen, T.; Vuolteenaho, K.; Kautiainen, H.; Nieminen, R.; Möttönen, T.; Hannonen, P.; Korpela, M.; Kauppi, M.J.; Laiho, K.; Kaipiainen-Seppänen, O.; et al. Glycoprotein YKL-40: A potential biomarker of disease activity in rheumatoid arthritis during intensive treatment with csDMARDs and infliximab. Evidence from the randomised controlled NEO-RACo trial. PLoS ONE 2017, 12, e0183294.
- Kazakova, M.H.; Pavlov, G.A.; Dichev, V.D.; Simitchiev, K.K.; Stefanov, C.S.; Sarafian, V.S. Relationship between YKL-40, neuron-specific enolase, tumor necrosis factor-a, interleukin-6, and clinical assessment scores in traumatic brain injury. Arch. Trauma Res. 2021, 10, 23.
- Magdalinou, N.K.; Paterson, R.W.; Schott, J.; Fox, N.; Mummery, C.; Blennow, K.; Bhatia, K.; Morris, H.; Giunti, P.; Warner, T.; et al. A panel of nine cerebrospinal fluid biomarkers may identify patients with atypical parkinsonian syndromes. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1240–1247.
- Hall, S.; Surova, Y.; Öhrfelt, A.; Blennow, K.; Zetterberg, H.; Hansson, O. Longitudinal Measurements of Cerebrospinal Fluid Biomarkers in Parkinson’s Disease. Mov. Disord. 2016, 31, 898–905.
- Olsson, B.; Constantinescu, R.; Holmberg, B.; Andreasen, N.; Blennow, K.; Zetterberg, H. The glial marker YKL-40 is decreased in synucleinopathies. Mov. Disord. 2013, 28, 1882–1885.
- Wennstrom, M.; Surova, Y.; Hall, S.; Nilsson, C.; Minthon, L.; Hansson, O.; Nielsen, H.M. The Inflammatory Marker YKL-40 Is Elevated in Cerebrospinal Fluid from Patients with Alzheimer’s but not Parkinson’s Disease or Dementia with Lewy Bodies. PLoS ONE 2015, 10, e0135458.
- Villar-Piqué, A.; Schmitz, M.; Hermann, P.; Goebel, S.; Bunck, T.; Varges, D.; Ferrer, I.; Riggert, J.; Llorens, F.; Zerr, I. Plasma YKL-40 in the spectrum of neurodegenerative dementia. J. Neuroinflamm. 2019, 16, 145.
More