The pathophysiological process of intracerebral hemorrhage (ICH) is very complex, involving various mechanisms such as apoptosis, oxidative stress and inflammation. As one of the key factors, the inflammatory response is responsible for the pathological process of acute brain injury and is associated with the prognosis of patients. Abnormal or dysregulated inflammatory responses after ICH can aggravate cell damage in the injured brain tissue. The NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome is a multiprotein complex distributed in the cytosol, which can be triggered by multiple signals. The NLRP3 inflammasome is activated after ICH, thus promoting neuroinflammation and aggravating brain edema.
- intracerebral hemorrhage
- secondary brain injury
- NLRP3 inflammasome
1. Introduction
2. Functions of the NLRP3 Inflammasome in ICH
2.1. Activation of the NLRP3 Inflammasome
The NLRP3 inflammasome consists of the effector protein pro-caspase-1, the adapter protein apoptosis-associated speck-like protein (ASC) and the sensor protein NLRP3 and orchestrates innate immune responses against cell stress and infection by regulating the caspase-1-dependent pathway and releasing proinflammatory cytokines such as interleukin-1β (IL-1β) and IL-18 [10][20]. The sensor protein NLRP3 consists of a carboxy-terminal leucine-rich repeat (LRR) domain, a NACHT domain and a pyrin domain (PYD). Upon binding to the corresponding ligands, thee LRR domain regulates the function of LRRs. The activation of the NLRP3 inflammasome involves two stages, i.e., the priming and activation stages (Figure 1). The priming stage is induced by the recognition of two molecular patterns, i.e., the damage-associated molecular patterns (DAMPs) and the pathogen-associated molecular patterns (PAMPs) [11][21]. This can activate the NF-κB signaling pathway and promote the expression of precursor proteins, including pro-IL-1β, pro-IL-18 and NLRP3. The activation stage is triggered by multiple stimuli that exist during metabolic imbalance, infection or tissue injury. When dangerous signals are recognized, the NLRP3 protein structure changes and exposes its PYD domain, which will bind to the PYD of ASC by forming a PYD–PYD interaction. Subsequently, ASC recruits the cysteine protease pro-caspase-1 to assemble the inflammasome complex via interacting with the caspase recruitment domain (CARD). Pro-caspase-1 is activated by self-cleavage to form active caspase-1. Then, caspase-1 dissociates gasdermin D (GSDMD) to release its N-terminal domain, which in turn binds to phosphatidylinositol phosphates and phosphatidylserine in the cytomembrane to generate pores, thereby inducing a lytic form of cell death, known as “pyroptosis”. In addition, caspase-1 can induce the transformation of IL-1β and IL-18 precursors into mature IL-1β and IL-18 and eventually aggravate the inflammatory responses and related complications.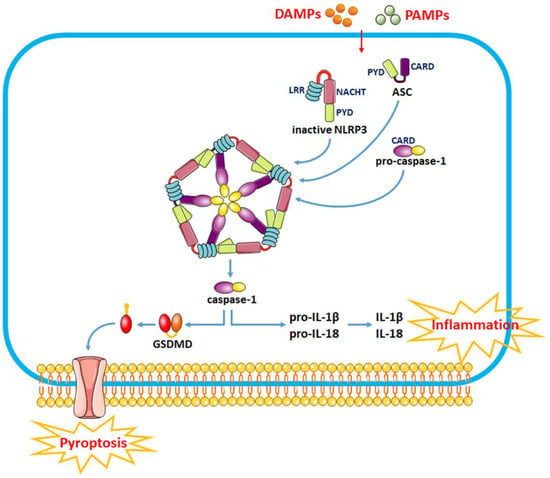
2.2. Modulation of NLRP3 Inflammasome Activity as a Therapeutic Strategy for SBI after ICH
The NLRP3 inflammasome is a multimolecular complex in the cytoplasm that mediates caspase-1 processing and proinflammatory cytokine maturation, including IL-1β and IL-18. Various risk factors such as Ca2+ mobilization, Na+ influx, K+ efflux, chloride efflux, mitochondrial dysfunction, oxidative stress and lysosomal damage are involved in the activation of the NLRP3 inflammasome to mediate neuroinflammatory responses after ICH [9][12][9,22]. Activating the NLRP3 inflammasome generates high levels of inflammatory cytokines, triggers an inflammatory response and recruits other immune cells to clear DAMPs after hemorrhage. However, overactivation of the NLRP3 inflammasome can result in persistent neuroinflammation and brain injury after ICH. Thus, assessing the role of the NLRP3 inflammasome in the processes associated with ICH may provide new strategies for ICH therapy. As a crucial component of innate immunity upon tissue injury, the NLRP3 inflammasome is activated after ICH, thereby promoting neuroinflammation and aggravating brain edema [8]. It has been reported that the expression of NLRP3 is gradually upregulated in the perihematoma tissue within 1–5 days after ICH, and the NLRP3 inflammasome is responsible for the complement-induced neuroinflammation, which eventually leads to abnormal neurological functions [9]. Activating the NLRP3 inflammasome can promote neuroinflammation via caspase-1 processing and IL-1β generation following ICH. Nevertheless, ICH-induced NLRP3 inflammasome activation can promote neutrophil infiltration, trigger the inflammatory response, impair neurological functions and aggravate brain edema after ICH [9]. In recent years, numerous studies have been conducted around the functions of the NLRP3 inflammasome in ICH. Brain injury induced by inflammation after ICH can be alleviated by directly or indirectly inhibiting NLRP3 inflammasome activation. For example, histone deacetylase 10 (HDAC10) downregulates protein tyrosine phosphatase nonreceptor type 22 (PTPN22) expression by binding to and deacetylating the PTPN22 promoter, which inhibits NLRP3 inflammasome activation and alleviates inflammation after ICH in rats. PTPN22 is a protein tyrosine phosphatase involved in the cellular immune response and inflammation and is related to various autoimmune diseases. PTPN22 binds and dephosphorylates NLRP3 following proinflammatory injury, thus promoting NLRP3 activation and IL-1β secretion. Interfering with PTPN22 can reduce the expression of IL-1β and IL-18 by inhibiting the activation of the NLRP3 inflammasome to reduce inflammatory responses, thereby improving neurological dysfunction and reducing cerebral edema in ICH rats [13][23]. Mammalian sterile-20-like kinase 4 (MST4) is a member of the glucokinase (GCK) subfamily, which directly phosphorylates TRAF6 to suppress inflammation. MST4 phosphorylates and activates TRAF6, which further activates the NLRP3 inflammasome by regulating the TLR/IL-1R signaling pathway [7]. Overexpression of MST4 negatively regulates the NLRP3 inflammasome and reduces the expression of tumor necrosis factor-α (TNF-α) and IL-1β, indicating that NLRP3 and MST4 may be potential therapeutic targets for neuroinflammation after ICH [7]. Mitochondrial dysfunction plays an essential role in the activation of the NLRP3 inflammasome [14][24]. Mitophagy is involved in the maintenance of mitochondrial homeostasis via selective degradation of damaged mitochondria, which prevents inflammation by inhibiting the NLRP3 inflammasome pathway [15][25]. Therefore, modulating mitophagy to inhibit NLRP3 inflammasome activation can become a therapeutic approach for alleviating secondary brain injury (SBI) after ICH. Chen et al. revealed that the activation of the Nrf2/Optineurin (OPTN) pathway can mediate mitophagy to alleviate SBI by suppressing the NLRP3 inflammasome following ICH. OPTN is a multifunctional ubiquitin-binding autophagy receptor that interacts with Nrf2 to mediate mitophagy and eliminate damaged mitochondria following ICH. Suppressing Nrf2/OPTN could enhance NLRP3 inflammasome activation, downregulate mitophagy levels and increase BBB disruption and brain edema, with more severe neurological deficits after ICH [16][26]. FUN14 domain-containing 1 (FUNDC1) is a mitophagy receptor that is overexpressed after ICH. FUN14 can suppress the NLRP3 inflammasome via regulation of mitophagy, thus alleviating ICH-induced injury. However, silencing FUNDC1 promotes NLRP3-mediated inflammation, thereby suppressing mitophagy and exacerbating ICH injury [17][27]. Thus, it can be seen that modulating mitophagy to inhibit NLRP3 inflammasome activation is an important therapeutic strategy for alleviating ICH injury. Many drugs can improve brain dysfunction and protect against SBI after ICH by acting NLRP3 inflammasome pathways (Table 1). For example, pioglitazone, edaravone and adiponectin significantly reduced brain edema and attenuated neurological deficits after ICH, as well as decreased the expression of IL-1β, IL-18, caspase-1 and NF-κB through suppressing the expression of NLRP3 [8][18][19][8,28,29]. Glibenclamide markedly reduced the neurological deficit and brain edema after ICH by decreasing the expression of ASC and caspase-1 and suppressing the activation of the NLRP3 inflammasome to maintain BBB integrity [20][30]. Memantine reduces ONOO- production by inhibiting neuronal nitric oxide synthase (nNOS) phosphorylation at ser1412, which further inhibits MMP-9 expression and NLRP3 inflammasome activation, protecting the blood–brain barrier integrity and alleviating neurological deficits in ICH rats [21][31]. Atorvastatin can protect the neurological function and reduce neuroinflammation and neuronal apoptosis by reducing the expression of TNF-α, IL-6 and IL-1β in ICH model mice. Furthermore, atorvastatin can decrease the expression of NLRP3 and cleaved caspase-1 and reverse the increase in toll-like receptor 4 (TLR4) and myeloid differentiation factor 88 (MyD88), indicating that atorvastatin suppresses NLRP3 inflammasome activation in glial cells of ICH model mice through inhibiting MyD88- and TLR4-associated pathways [22][32]. Some traditional Chinese medicine such as isoliquiritigenin, silymarin, baicalein and cordycepin can also exert antioxidant and anti-inflammatory roles by significantly inhibiting the activation of the Nrf2 dependent-NF-κB pathway and NLRP3 inflammasome, which in turn mitigates brain edema and improves neurological deficits after ICH [23][24][25][26][27][33,34,35,36,37]. This evidence indicates that the suppression of NLRP3 inflammasome activation might be a therapeutic target for ICH recovery.|
Drugs |
Models |
Efficacy |
References |
|---|---|---|---|
|
Pioglitazone |
blood-induced mouse ICH model |
brain edema↓, lactate↑ |
|
|
Edaravone |
autologous blood-induced rat ICH model |
IL-1β↓, caspase-1↓, NF-κB↓, brain edema↓, neurological deficits↓ |
|
|
Adiponectin |
autologous blood-induced rat ICH model |
IL-1β↓, IL-18↓, brain edema↓, neurological deficits↓ |
[8] |
|
Glibenclamide |
autologous blood-induced mouse ICH model |
IL-1β↓, IL-18↓, IL-6↓, TNF-α↓, brain edema↓, disrupted BBB↓, neurological deficits↓ |
|
|
Memantine |
collagenase-induced rat ICH model |
IL-1β↓, disrupted BBB↓, neurological deficits↓ |
|
|
Atorvastatin |
collagenase-induced mouse ICH model |
IL-1β↓, IL-6↓, TNF-α↓, brain edema↓, neurological deficits↓ |
|
|
Isoliquiritigenin |
collagenase IV-induced rat ICH model |
NF-κB↓, IL-1β↓, brain edema↓, disrupted BBB↓, neurological deficits↓ |
|
|
Silymarin |
collagenase II-induced mouse ICH model |
NF-κB↓, caspase-1↓, IL-1β↓ |
|
|
Baicalein |
collagenase VII-induced rat ICH model |
ROS↓, SOD↑, GSH-Px↑, ASC↓, caspase-1↓ |
|
|
Cordycepin |
autologous blood-induced mouse ICH model |
IL-1β↓, IL-18↓, brain edema↓, neurological deficits↓ |