Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Bryan Mathis and Version 2 by Dean Liu.
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a master regulator of the endogenous antioxidant response to reactive oxygen species as well as a controller of Phase II detoxification in response to xenobiotics. However, more is not necessarily better as the heart's unique biochemical and molecular composition make it susceptible to damage if the Nrf2 response is uncontrolled.
- Nrf2
- Keap1
- cardiomyopathy
- bardoloxone
1. Introduction
Reactive oxygen species (ROS) are both a normal byproduct of mitochondrial metabolism and an endproduct of oxidative biochemical reactions in the cell. Balanced levels of subcellular compartmental ROS are important for normal cellular functions, whereas dysregulated ROS, usually caused by relative insufficiency or impairment of the endogenous antioxidant defense system, attack cellular components leading to cellular damage and death, a state referred as to oxidative stress. To maintain cellular redox homeostasis and neutralize uncontrolled ROS, conserved antioxidant defense enzymes are placed under the control of the nuclear factor erythroid 2-related factor 2 (Nrf2) gene which is constitutively expressed in all higher-order animals. This gene, as a master antioxidant transcription factor, is responsible for global antioxidant activity in response to internally and externally sourced ROS threats but also modulates such species to maintain important intracellular second messenger capability. As the inhibitor of Nrf2, Kelch-like erythroid cell-derived protein with CNC homology-associated protein 1 (Keap1), is amenable to attack by exogenous compounds, research has focused its efforts to increase Nrf2 activity via direct interaction with Keap1. These compounds, such as oleanic acid derivative 2-cyano-3,12-dioxoolean-1,9- dien-28-oic acid (CDDO, bardoloxone), have been intensely studied in animal models and human trials as potential defense agents oxidative stress-associated diseases, such as cancer, chronic kidney disease, fatty liver, and endometriosis. Unfortunately, human trials employing CDDO and similar compounds for amelioration of these maladies have met with ambiguous and often disappointing results. Furthermore, multiple trials throughout 2007–2014 were terminated as unforeseen myocardial pathologies resulted. What follows is a survey of Nrf2, its endogenous regulation, action, and potential for exogenous modulation. Additionally, current clinical trial results are presented and analyzed for characteristics of Nrf2 upregulation that result in possible cardiac issues. Finally, comparisons between these pharmaceutical regulators and exercise are made from mechanistic and physiological viewpoints to elucidate the differences between endogenous and exogenous regulatory effects on Nrf2 and cardiac health.
2. Nrf2 in the Myocardium: Not a Silver Bullet
The heart is obligately aerobic and relies on oxidative phosphorylation to generate the biochemical energy needed for a lifetime of pumping. The coronary arteries supply oxygenated blood to the heart during diastole and increases in oxygen demand by the myocardium are directly related to the heart rate (higher rate = higher oxygen demand and shorter diastole for coronary supply) and saturation of blood by oxygen (to prevent hypoxia). Even at rest, the myocardium consumes 8 to 13 mL of oxygen per 100 g of tissue per minute and ROS from mitochondrial respiration and pro-ROS proteins, such as Nox4, create a pro-oxidative state that requires constant rebalancing to maintain redox homeostasis [1][2][48,49]). Xenobiotics may also introduce ROS either by direct chemical action (e.g., nitrosamines from cigarette smoke, fermented foods, or cured meats) or immune response. However, since ROS function as a second-messenger system and have been implicated as crucial regulators of stem cell differentiation and apoptosis/necrosis, tight regulation of the Nrf2-mediated antioxidant response (e.g., via Keap1 direct and Bach1 competitive pathways) is required to maintain such basal messenger activity. Cardiomyocyte differentiation, in particular, is sensitive to ROS, requiring it for progression to maturity, and cardiac-resident stem cells in adults may be similarly affected by imbalanced redox homeostasis, driving them towards hypertrophic or synthetic phenotypes [3][50]. Of current controversy in cardiac research is the involvement of Nrf2 as a pro-hypertrophic, factor in progressive heart failure. On one side, numerous reports have linked Nrf2 deficiencies to ROS-mediated cardiac hypertrophy related to Angiotensin II, IL-6-mediated inflammation, aortic constriction (TGFβ1/SMAD2 signaling), and obesity-related stress [4][5][6][51,52,53]. Diverse other reports have detailed the role of Nrf2 in preventing cardiomyocyte necrosis, hypertrophy, and fibrosis of the myocardium due to ROS while antioxidant response proteins (e.g., NQO1, SOD1, GPX4) have been found at low expression levels under ischemic cardiomyopathy conditions [7][8][54,55]). However, recent evidence that Nrf2 induces progressively maladapted remodeling in the absence of functional autophagy casts doubt on the exploitation of Nrf2 in patients with metabolic disorders or heart disease (Figure 1) [9][46]. Reports from the Cui research group have indicated that Fyn-mediated nuclear export inhibition is to blame but other yet-discovered factors may also play crucial roles in pathogenesis [9][46]. Future studies on the effect of autophagy and other regulatory modalities (methylation, sumoylation, etc.) will delineate the thresholds beyond which Nrf2 enhancement becomes problematic for the heart. Ostensibly, boosting Nrf2 will increase the total antioxidant capacity within the heart and neutralize ROS that perpetuate necrotic and fibrotic pathways, leading to the concept of “the more antioxidant capacity, the better”. In spite of this theory, results from well-controlled clinical trials of supplemental antioxidants (selenium, vitamin E, beta-carotene, etc.) have returned disappointing results where risk was either unchanged or even enhanced [10][56]. Results from previously reviewed meta-studies with 156,663 and 188,209 total participants found no significant effects of antioxidant/vitamin supplements on cardiovascular risk [10][56]). However, a recent meta-study of selenium and other antioxidants only found significant risk reduction for selenium across 43 studies (possibly because such minerals, similarly to zinc, are important constituents of antioxidant enzymes and not activators of Nrf2) [11][57]). Consequently, the Selenium and Vitamin E Cancer Prevention trial (N = 35,533) found that supplementation increased diabetes and prostate cancer risks, while a beta-carotene study did find inverse relationships with lower cardiovascular risk but could not completely rule out the effects of confounding variables (i.e., accidents and injuries) [11][12][57,58]. In general, antioxidants have proven to be poor substitutes for generally healthy lifestyle habits (e.g., no tobacco use, moderate diet, moderate exercise, stress reduction, good sleep habits) and excessive antioxidant use is associated with increased all-cause mortality (vitamin E), oxidative stress (ascorbic acid), and cancer risk (vitamin A) [13][59]. In similar fashion, Nrf2 exogenous enhancers have not shown promise in either preventing or treating cardiovascular diseases and several trials have ended early because of deleterious heart effects after treatment. For this reason, external and sustained enhancement of the antioxidant response out of context with other regulatory factors (e.g., autophagy) could counterintuitively damage the myocardium through pathways not yet fully elucidated (Figure 1). More antioxidant capacity is, in light of these studies, definitely not better.3. Nrf2 in the Failing Heart: Autophagy as a Keystone Mechanism
Aging and failing hearts experience stiffening from fibrosis caused by immune responses to myocardial necrosis, increased ROS from aging and senescent mitochondria, lipofuscin accumulation from lysosomal degradation, deficiencies in calmodulin signaling/calcium flux (RYR2, SERCA2a) and increased maladaptive remodeling due to high blood pressure that stems from glucose dysregulation and hyperkalemia [14][15][16][60,61,62]. Additionally, autophagic capacity drops as suppression factors like mTOR are overexpressed by chronically high AKT levels while chronic IGF-1 expression, long touted as a youth-sustaining factor, paradoxically ages the heart rapidly as it has been shown to downregulate autophagy by suppression of autophagosome formation and increases in AKT/mTOR [17][18][63,64]. Hyperglycemia has been shown to modulate autophagy via AMPK and ROS induction of the ERK/JNK-p53 mechanism [19][20][21][65,66,67]. Additionally, fasting is a potent activator of autophagy even under increased peroxide generation by mitochondria in animals [20][22][66,68]. In type 2 diabetics, while initially protective, mitophagy (i.e., autophagy of damaged mitochondria) may eventually drive cells towards reduced energy as mitochondria are damaged by increased metabolic activity and are recycled faster than replacement [23][69]. However, the loss of autophagic capacity, especially in pancreatic β cells and diabetic hearts, may also be important in progression to end-stage disease [23][24][69,70]). Thus, patients who do not possess a fully intact autophagy capacity (e.g., heart failure or type 2 diabetics) may be harmed by artificial Nrf2 enhancement. Wu et al. recently reported a putative mechanism for this effect in pressure-overloaded hearts that involves dysfunctional autophagy, restricting phosphorylated Fyn and ERK from translocating to the nucleus and downregulating Nrf2 activity that would otherwise restrict angiotensin expression [25][71]. In such cases, subsequent activation of angiotensin II (Ang-II) receptors by Ang-II production would increase blood pressure and eventual hypertrophy [25][71]. Additionally, interactions between autophagic control factor p62 and Keap1 mean that reduction in upstream p62/AKT/mTOR result in increased Nrf2 activation and further exacerbation of Ang-II-induced maladaptive remodeling (Figure 1) [26][72].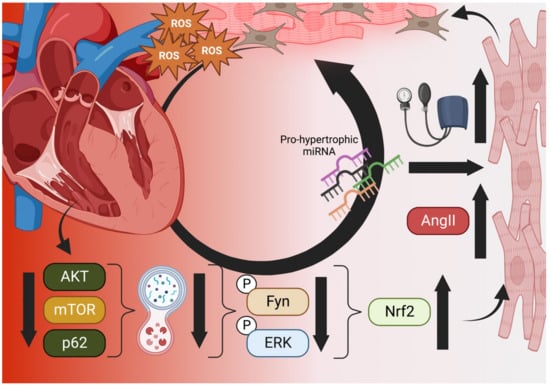
Figure 1. The Vicious Cycle of Nrf2 in Cardiac Hypertrophy. Aged and failing hearts have dysfunctional autophagy (bottom), which cannot downregulate Nrf2 transcription of Angiotensinogen and AngII, increasing blood pressure and mechanical induction of hypertrophy, pro-hypertrophic miRNA, necrosis, and fibroblast activation (right). Hypertrophic cells increase ROS output and decompensation within the heart occurs, increasing the ischemic microenvironment and generating even more ROS in a vicious cycle (top) [25][26][71,72]. Created in BioRender.com.