Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Priscilla Cascetta and Version 3 by Dean Liu.
In NSCLC (Non-Small Cell Lung Cancer), KRAS (Kirsten Rat sarcoma virus) mutations occur in up to 30% of all cases, most frequently at codon 12 and 13. KRAS mutations have been linked to adenocarcinoma histology, positive smoking history, and Caucasian ethnicity, although differences have been described across KRAS mutational variants subtypes. KRAS mutations often concur with other molecular alterations, notably TP53, STK11, and KEAP1, which could play an important role in treatment efficacy and patient outcomes.
- KRAS
- NSCLC
- lung cancer
- adagrasib
1. Introduction
With almost 1.8 million deaths per year, lung cancer remains the leading cause of cancer-related death worldwide. Non-small cell lung cancer (NSCLC) accounts for 85% of all diagnosed cases, with adenocarcinoma being the most common histological subtype [1][2][1,2]. Progress in molecular characterization and development of new targeted therapies have changed the treatment scenario of NSCLC in recent years. So far, molecular alterations thought to be undruggable for many years, such as KRAS mutations, may now benefit from targeted agents.
KRAS (Kirsten Rat sarcoma virus) proteins are small GTP-ases belonging to the wider family of RAS proteins. The KRAS gene is located in the short arm of chromosome 12 (12 p) and its transcripts may further undergo an alternative splicing process, resulting in two distinct proteins of approximately 21 KDa each: KRAS 4A and KRAS 4B [3]. Even though differences in physiological expression have been reported in some preclinical data [4], little is known about the real impact of these alternatively spliced variants in NSCLC.
Structurally, all RAS isoforms have a highly conserved catalytic region, implied in GTP bounding and hydrolysis. Differences across RAS isoforms are mainly founded at the C-terminal hyper variable region (HVR) domain, responsible for post-transcriptional modifications and interaction with the phospholipidic membrane bilayer [5][6][5,6].
In physiological conditions, KRAS proteins are key elements in transducing signaling from activated receptor tyrosine kinase (RTK) to downstream intracellular pathways. Indeed, intracellular KRAS proteins may present in two distinct functional forms: KRAS-GTP and KRAS-GDP, which correspond to “active” and “inactive” states, respectively. Transition from the inactive state (KRAS-GDP) to active state (KRAS-GTP) is usually mediated by the guanine nucleotide exchange factors (GEFs) via the releasing of guanosine diphosphate (GDP) and the subsequent binding of guanosine triphosphate (GTP). Many GEFs proteins have been identified to date, with the GEF Son of Sevenless (SOS1) playing a crucial role in KRAS activation. Of note, this process determines key structural modification in KRAS proteins mainly involving the switch pockets I and II, which serve as the binding interface for effector proteins [7][8][7,8]. However, the transition from the inactive to active form is complex and usually requires other proteins (e.g., the adaptor protein Grb2 and the SOS1/Grb2 recruiter SHP-2) which directly interact with upstream phosphorylated RTKs [9]. Contrarily, transition from the KRAS-GTP to KRAS-GDP binding state is facilitated by GTPase activating proteins (GAPs) such as NF1, which accelerate the hydrolysis of GTP to GDP [10].
Downstream pathways of KRAS normally include the RAF-MEK-ERK mitogen-activated protein kinase (MAPK) pathway, the phosphatidylinositol 3-kinase (PI3K)-AKT-mTOR, and the RAS-like (RAL) pathways [5], whose deregulations have been linked with abnormal cellular proliferation, migration, and death (Figure 1).
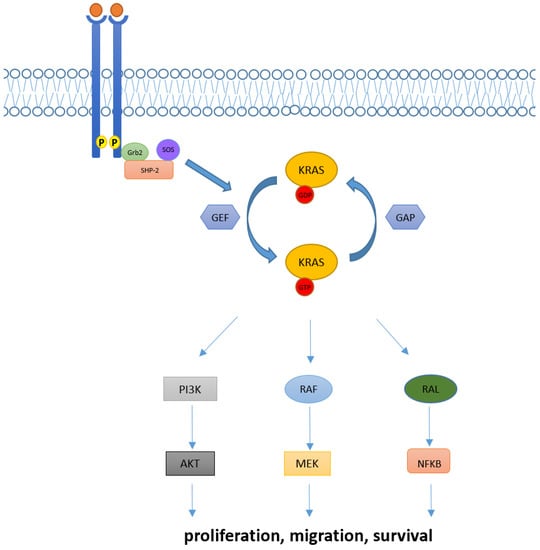
Figure 1. KRAS activation and its downstream pathway. KRAS proteins are key elements in transducing extracellular signaling to downstream pathways. Intracellular KRAS proteins may present in two functional forms, KRAS-GTP and KRAS-GDP, which correspond to their active and inactive state, respectively. Transition from the KRAS-GDP to KRAS-GTP binding state is usually mediated by GEFs proteins and occurs via other adaptor and recruiter proteins, such as Grb2 and SHP-2. Conversely, transition from KRAS-GDP to KRAS-GTP usually requires GAPs proteins. Downstream signaling of KRAS normally includes RAF-MEK-ERK, PI3K/AKT and RAL pathways, implied in the regulation of cellular proliferation, migration, and death.
The curesearchers rent review provides an overview of the biology and clinic-pathological characteristics of KRAS mutant NSCLC patients. Moreover, researcherswe discuss therapeutic strategies that have been explored in these subgroups of patients and the clinical data leading to the approval of sotorasib and adagrasib, the first compounds that efficiently inhibit KRAS G12C mutation. ResearchersWe then focus on acquired resistance mechanisms evidenced to date and researcherswe further assess the role of immune checkpoint inhibitors in KRAS mutant patients. Finally, researchers we look into ongoing trials and new KRAS inhibitors, which are currently being tested.
2. Techniques for Detection of KRAS Mutations
The assessment of the KRAS status in patients affected by colorectal cancer and NSCLC is mandatory for a correct therapeutical management [11]. The main techniques allowing KRAS point mutations are polymerase chain reaction (PCR) based methods, such as high-resolution melting and real time RCP (RT.-PCR), and next generation sequencing (NGS) [12][13][14][12,13,14]. RT-PCR allows the highly sensitive qualitative detection and identification of a fixed number of mutations in specific areas of the gene [13]. Highly verified assays, such as Cobas Mutation Test V2, reaches a 100% level of accuracy in tissue samples, analyzing exons 2, 3, and 4 of the KRAS gene [15]. NGS technologies, being DNA-based or RNA-based, enable the simultaneous analysis of a high variety of genes and loci, at the price of reducing sensitivity, when compared to PCR [16]. With the advent of new molecular targets, an exhaustive characterization of lung cancer patients has become essential, making NGS an extremely valuable tool [17][18][19][20][17,18,19,20].
The sensitivity of KRAS mutation detection has been evaluated and compared in various types of specimens, such as tissue, plasma, and other body fluids, finding a high rate of concordance between tissue and liquid biopsy both for PCR and NGS [21][22][21,22]. The use of liquid biopsy is of great relevance for the management of advanced NSCLC, enabling the identification of KRAS mutations at the baseline, but also to picture disease heterogeneity, to evaluate response to treatment in time and to identify molecular mechanisms of resistance to targeted molecules [22][23][24][22,23,24]. Overall, tissue-based testing remains the preferred test for most cancer patients, although liquid biopsy may be chosen when faster results will be clinically important, or when obtaining tissue specimens is not possible or inappropriate [23] However, it is important to point out that reimbursement for broad coverage is still limited [25].
3. KRAS in NSCLC: Different Alterations and Patients’ Characteristics
In NSCLC, KRAS mutations are founded in up to 30% of all cases and most frequently involve the codon 12 (90% of cases), while less common mutations are observed in codon 13 (2–6%) and 61 (1%) [26][27][26,27]. Within codon 12 mutations, the one resulting in the substitution of glycine with cysteine (KRAS G12C) represents the most prevalent KRAS alteration in NSCLC (40% of all KRAS mutant cases), followed by substitution of glycine with valine (KRAS G12V, 21%), and substitution of glycine with aspartic acid (KRAS G12D, 17%). Indeed, other point mutation in codon 12 such as G12A/R/S are rare [26][27][28][26,27,28]. Functionally, mutations at codon 12 and 13 result in reduced GAP-mediated hydrolysis via allosteric bloc that hinders the GTP-ase site of KRAS, whereas alterations in codon 61 abolish both intrinsic and GAP-mediated GTP hydrolysis of KRAS [29]. Of note, KRAS mutant variants can differently activate their downstream pathway. Indeed, the hydrophobic G12C and G12V preferentially activate the RAL pathway, while the hydrophilic G12D mainly acts via PI3K-AKT signaling. KRAS G12C weakens the interaction with PI3K, and the bulky aspartic acid in the G12D prevents the formation of the binding with RelA/RelB [30]. Besides NSCLC, KRAS mutations are also frequent in other tumor types, such as pancreatic and colorectal adenocarcinoma (88% and 50% of all cases, respectively) [28][31][28,31], with KRAS G12D mutations being the most common alteration among these histotypes (32% and 46% of all cases, respectively) [32].
In lung cancer patients, KRAS mutations have been associated with Caucasian ethnicity (Caucasian vs. Asian population: 26% vs. 11%), female sex (female vs. males: 31.35 vs. 23.7%, p < 0.0001), adenocarcinoma histology (adenocarcinoma vs. squamous cell carcinoma: 37.2% vs. 4.4%), and a positive smoking history (smokers vs. non-smokers: 30% vs. 11%) [26][28][26,28]. However, these characteristics may strongly depend on single mutant KRAS variants. As such, KRAS G12C/G12A/G12V/G13C mutations are more frequently found in current or former smokers, while prevalence of KRAS G12D/G12S/G13D mutations is higher among non-smokers [26][33][26,33]. Furthermore, the median age for KRAS G12C patients seems to be significantly lower than that observed for other KRAS mutants (KRAS G12C vs. other KRAS-MUT: 63.1 vs. 65.9 years, p = 0.0092) [34], although this evidence has not been further confirmed in other reports [26]. Finally, the metastatic pattern could also slightly differ across single KRAS mutant variants, since KRAS G12C mutants are more likely associated with lung metastasis (KRAS G12C vs. non–G12C: 38% vs. 21%; p = 0.043) and less associated with either pleural metastasis or lymphangitic carcinomatosis (KRAS G12C vs. non–G12C: 4% vs. 39%, p = 0.0001) [35]. Of note, KRAS mutant patients do not have a higher brain tropism than those without KRAS alterations, and the same incidence of brain metastasis has been observed across different KRAS mutant variants (KRAS mut vs. KRAS wild-type: 33% vs. 40%, p = 0.17; KRAS G12C vs. KRAS other: 40% vs. 41%, p = 0.74) [36].
4. Predictive and Prognostic Role of KRAS Mutations in NSCLC Patients
Multiple studies have evaluated the predictive and prognostic role of KRAS mutations in NSCLC.
In an early setting, different reports have demonstrated similar overall survival (OS) and disease-free survival (DFS) in KRAS mutants versus wild type patients (OS HR = 1.17, DFS HR = 1.04) [37][38][37,38]. However, these results were not further confirmed and other reports eventually demonstrated a worse OS and DFS for KRAS mutant patients (DFS p = 0.038, OS HR = 1.30; p = 0.002) [37].
It has been asked if the prognostic and predictive significance of KRAS mutation could depend on specific variants. Indeed, Finn et al. recently demonstrated a negative prognostic impact for KRAS G12C completely resected patients compared to both the KRAS wild type and other mutant variants (OS KRAS G12C versus other KRAS HR = 1.39, p = 0.031; KRAS G12C versus no KRAS HR = 1.32, p = 0.028) [34]. Contrarily, no prognostic nor predictive value for codon 12 mutations has been identified in a pooled meta-analysis conducted in early stage disease, while codon 13 mutations have been associated with a possible deleterious effect to adjuvant chemotherapy [37].
In an advanced setting, a meta-analysis of 43 selected studies demonstrated an impaired OS and progression-free survival (PFS) for KRAS mutants (OS HR = 1.71; 95% CI [1.07, 2.84]; PFS HR = 1.18; 95% CI [1.02, 1.36]). However, these results were only observed when considering observational studies and not further confirmed in randomized clinical trials (OS HR = 1.10; 95% CI [0.88–1.38]; PFS HR = 1.03; 95% CI [0.80–1.33]) [39]. Likewise, discordant results about the prognostic and predictive role of KRAS mutations have been reported in the advanced setting [27][40][41][27,40,41]. Furthermore, no differences in OS have been observed across different KRAS mutations, although a better response to taxanes has been reported for KRAS G12V mutants [33][42][33,42].
In conclusion, the prognostic and predictive role of KRAS mutations in NSCLC is still a matter of debate and discordant results provided so far need further assessment.
5. Concurrent Molecular Alterations in KRAS Positive NSCLC Patients
Concurrent molecular alterations are often found in KRAS mutant NSCLC patients. In the largest cohort available to date, Riely et al. described a median of seven concomitant mutations in KRAS positive NSCLC patients, the most frequent being TP53 (39%), STK11 (30%), KEAP1 (24%), RBM10 (15%), and PTPRD (15%). Curiously, those harboring concomitant mutations in either STK11 or KEAP1 had a shorter OS (STK11: HR = 2, p = 0.006; KEAP1: HR = 2.8, p < 0.001) and the negative prognostic impact of STK11 and KEAP1 co-alterations has been further confirmed in multiple studies [43][44][43,44]. Of note, neither the TP53 status nor number of concurrent mutations affected OS [45]. In another retrospective study, Passaro et al. demonstrated that 77% of patients with KRAS mutations had a concurrent molecular alteration. Interestingly, the percentage of concomitant alterations did not differ between G12C and non G-12C KRAS mutants. Again, the most frequently reported co-alteration was TP53 (23%) followed by STK11 (15%). Importantly, alterations in CDK2/4 and receptor tyrosine-kinase (RTK) have also been described in KRAS mutant cases [46].
Importantly, concurrent alterations might affect the tumor microenvironment with consequent alterations in the immunogenic phenotype. As such, Skoulidis et al. demonstrated a lower immunogenic infiltrate in STK11 positive tumors, while the opposite was seen for concomitant TP53-mutated tumors [47].
It has also been demonstrated that co-mutations could differentially activate downstream pathways. Indeed, engineered mice harboring KRAS G12D alone or with concomitant TP53 or STK11 mutations, respectively, hyper-activated MEK/ERK or AKT/SRC pathways [48].
Concurrent alterations with other key oncogenic drivers have also been described. Of note, KRAS mutations are found in approximately 3% of MET ex14 alterations [49][50][49,50], and a few cases of concomitant KRAS/ALK and KRAS/ROS1 alterations have been reported [51][52][51,52]. Interestingly, KRAS mutations have been found as mechanisms of resistance to the first, second, and third generation of EGFR TKI [53][54][55][56][53,54,55,56].