Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Myeong-Sok Lee.
Cancer has a negative impact on patients’ health and livelihoods, making it one of the most formidable adversaries of humanity. Cancer immunoediting is a major mechanism for controlling cancer that consists of three sequential phases: elimination, equilibrium, and escape. The interactions between immune systems and cancer cells are dynamic and constantly evolve because of the surrounding living environment, medical interventions, or genetic factors.
- cancer resistance
- immunotherapy
- mechanism
1. Introduction
Cancer has a negative impact on patients’ health and livelihoods, making it one of the most formidable adversaries of humanity. Cancer immunoediting is a major mechanism for controlling cancer that consists of three sequential phases: elimination, equilibrium, and escape [1]. The interactions between immune systems and cancer cells are dynamic and constantly evolve because of the surrounding living environment, medical interventions, or genetic factors. These factors influence anti-tumor immune responses to varying degrees and at the same time, resistance mechanisms would most likely emerge [2]. The immune system’s role as a major driver of tumor evolution is based on the selective pressure exerted on cells that give rise to tumors [2,3][2][3].
Immunotherapy has been widely recognized as a breakthrough treatment for a number of malignancies, earning Drs. James P. Allison and Tasuku Honjo the Nobel Prize in Physiology or Medicine in 2018 for their pioneering work—discovery of a novel cancer therapy by inhibition of the brakes on the immune system [6,7][4][5]. Immune-based cancer therapy consists of two major approaches including immune checkpoint blockade (ICB) and adoptive T cell therapy (ACT). Several immune checkpoint inhibitors (ICIs) have been studied extensively [8,9][6][7]. ICB is best-known for its remarkable efficacy in treating melanoma, with approximately 20% of recipients experiencing a complete response. More importantly, immune checkpoint therapy also induces a durable complete remission of melanoma [10][8]. While immune related toxicities induced by PD-1 blockade is similar to those induced by CTLA-4 blockade, they are less common possibly due to PD-1/PD-L1 checkpoint may involve later in T cell response [11][9].
Tumor-infiltrating lymphocyte (TIL) therapy and chimeric antigen receptor (CAR) T-cell therapy are the two types of ACT-based therapy. Cell-based immunotherapy is based on the notion of using the cells of immune system to eliminate cancer [2]. TILs are prepared by isolating cells from surgical samples; the isolated TILs are then cultured to increase the number enough to be transfused back to the patients whose samples were collected.
2. Molecular Mechanisms
2.1. Tumor Intrinsic Factors of Resistance to Immunotherapy
Cancer cells employ sophisticated strategies such as avoiding immune recognition and inducing immunosuppressive TME to evade immune attack. These mechanisms may occur before (primary resistance) or after (adaptive resistance) immunotherapy. Several important tumor-intrinsic pathways are associated with primary and adaptive resistance including (1) the mitogen-activated protein kinase (MAPK) pathway and/or loss of phosphatase and tensin homolog (PTEN) expression, which enhances PI3K signaling, (2) expression of WNT/β-catenin signaling pathway, (3) loss of interferon-gamma (IFNγ) signaling pathways, and (4) loss of tumor antigen expression (Figure 1B) [10].
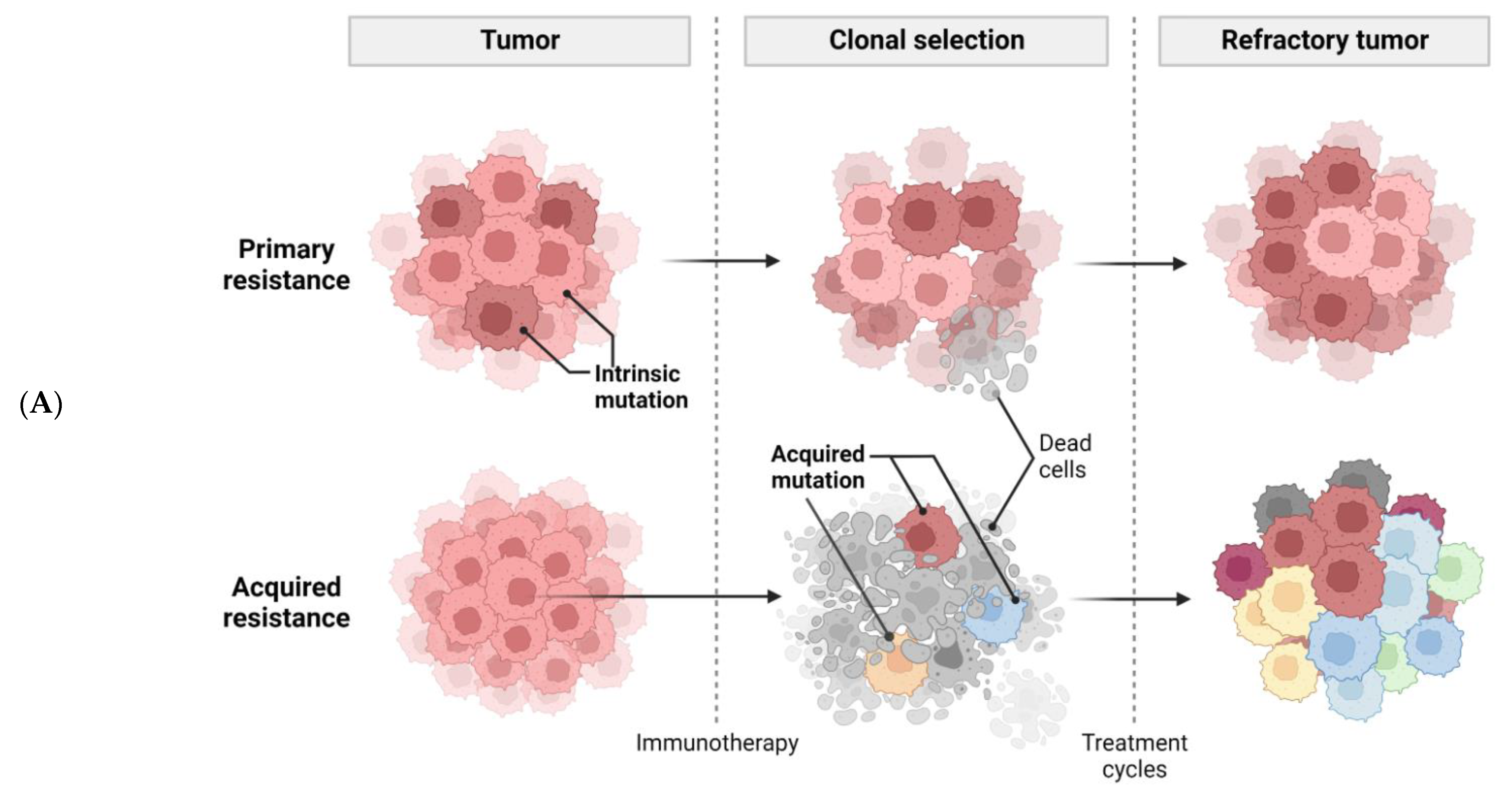
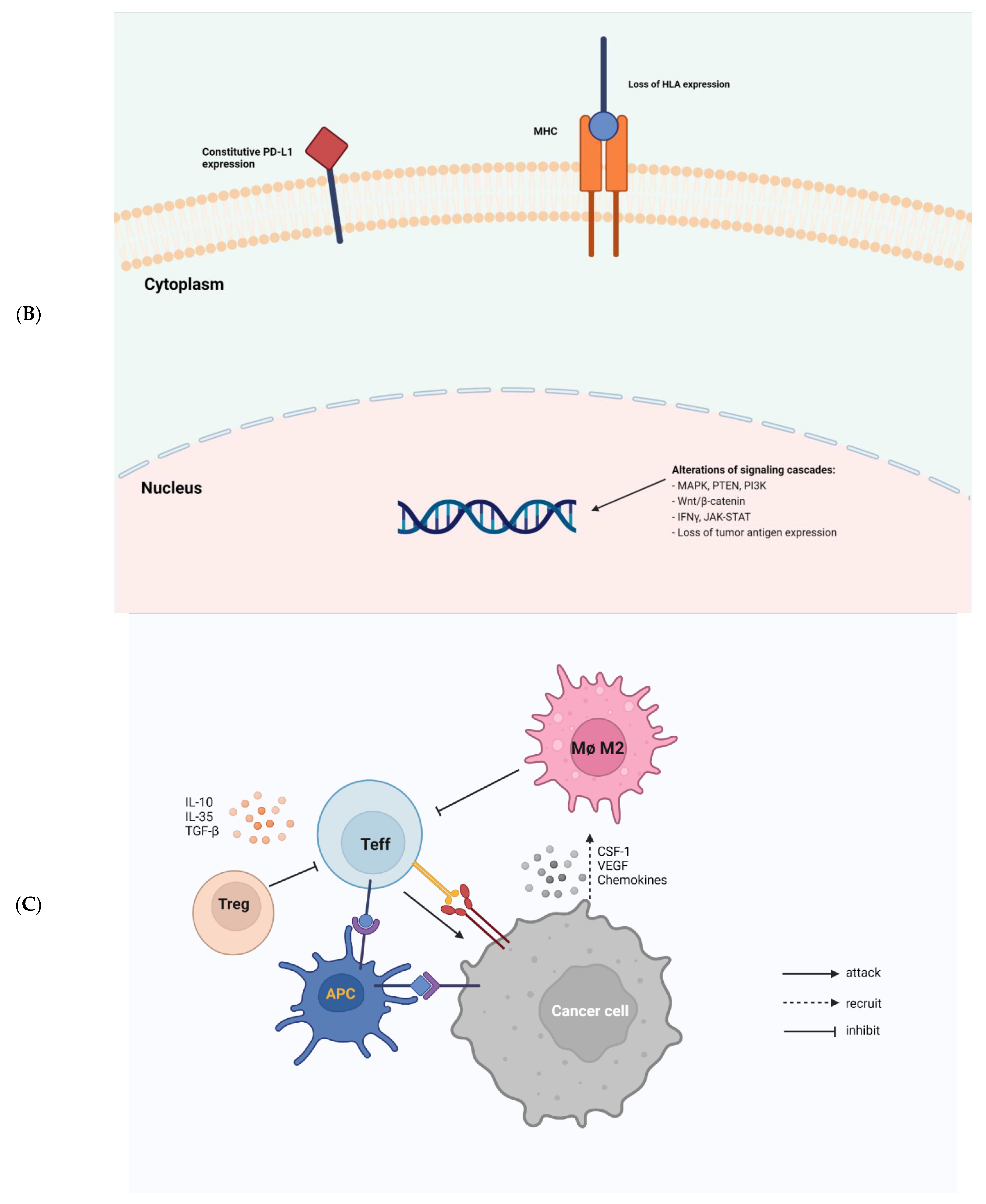 Figure 1. Tumor-intrinsic [25]and -extrinsic mechanisms of resistance to immunotherapy. (A), a general diagram of how cancer resistance emerges upon immunotherapeutic treatment. Intrinsic mutation in tumor cells renders cancer cells primary resistance upon immunotherapy leading to refractory tumor. Acquired resistance is generated in survived cancer cells upon immunotherapy leading to refractory tumor. (B), tumor-intrinsic mechanisms of resistance. Intrinsic factors include constitutive PD-L1 expression, loss of HLA expression in cancer cell membrane, and alterations of signaling cascades such as MAPK, PTEN, PI3K, Wnt/β-catenin, IFNγ, JAK-STAT, and loss of tumor antigen expression. (C), tumor-extrinsic mechanisms of resistance. The complex interplay of immune cells and cancer cells in tumor-microenvironment. Cancer cells are constantly subjected to bombardment by immune cells such as NKs and Teffs. However, cancer cells produce CSF-1, VEGF, and several chemokines to recruit M2 macrophages which in turn inhibit Teffs. Treg also induce cytokines, such as IL-10, IL-35, TGF-β to impede Teff responses.
Figure 1. Tumor-intrinsic [25]and -extrinsic mechanisms of resistance to immunotherapy. (A), a general diagram of how cancer resistance emerges upon immunotherapeutic treatment. Intrinsic mutation in tumor cells renders cancer cells primary resistance upon immunotherapy leading to refractory tumor. Acquired resistance is generated in survived cancer cells upon immunotherapy leading to refractory tumor. (B), tumor-intrinsic mechanisms of resistance. Intrinsic factors include constitutive PD-L1 expression, loss of HLA expression in cancer cell membrane, and alterations of signaling cascades such as MAPK, PTEN, PI3K, Wnt/β-catenin, IFNγ, JAK-STAT, and loss of tumor antigen expression. (C), tumor-extrinsic mechanisms of resistance. The complex interplay of immune cells and cancer cells in tumor-microenvironment. Cancer cells are constantly subjected to bombardment by immune cells such as NKs and Teffs. However, cancer cells produce CSF-1, VEGF, and several chemokines to recruit M2 macrophages which in turn inhibit Teffs. Treg also induce cytokines, such as IL-10, IL-35, TGF-β to impede Teff responses.In cancer, VEGF and IL-8 are induced by the MAPK cascade to inhibit the recruitment and functions of T cells [26][11]. Inhibition of MAPK signaling combined with PD-1/PD-L1-targeted or BRAF-targeted therapies leads to enhanced anti-tumor immune responses such as the increased presence of TILs [27,28,29][12][13][14]. Resistance to ICI can also be caused by the loss of PTEN, which promotes PI3K signaling to enhance proliferation of malignant cells [30][15]. PTEN loss in melanoma is associated with decreased gene expression of IFNγ and granzyme B in immune cells, as well as the infiltration of CD8+ T cells. PTEN deletions and mutations are more common in non-T-cell-inflamed tumors than in T-cell-inflamed tumors. As a result, ACT was less effective in treating PTEN-deficient tumors than PTEN-expressing tumors in mice model [30][15]. Moreover, a significant enrichment of somatic PTEN mutations was associated with resistance to ICIs, probably contributing to the immunosuppression environment in non-responders with glioblastomas [31][16].
The observation in many human cancers of an aberrant activation in the Wnt/β-catenin signaling has prompted the use of Wnt signaling inhibitors in cancer treatment. Constitutive Wnt signaling via β-catenin stabilization can induce T cell exclusion from the vicinity of cancer cells, impairing anti-tumor immunity and promoting immunotherapy resistance [32][17]. Experimental data from mice model suggest that tumors with elevated β-catenin levels have a loss of CD103+ DCs due to decreased CCL4 expression. Furthermore, ICB is more effective in targeting β-catenin loss tumors than β-catenin-expressing tumors. In the same vein, β-catenin signaling-related genes are higher expressed in non-T-cell-inflamed tumors [32][17]. Results from the clinical study by Luke et al. [33][18] strongly support the rationale of targeting Wnt/β-catenin pathway in cancer. Additionally, mutations in β-catenin signaling molecules showed a significant enrichment in non-T-cell-inflamed tumors, and moreover, activation of Wnt/β-catenin signaling was found in 90% of examined non-T-cell-inflamed tumors [33][18].
The IFNγ signaling pathway, which functions through JAK-STAT signaling [34][19], has dual effects on anti-tumor immunity. Tumor-specific T-cell-induced IFNγ can induce lethal anti-tumor immunity by promoting tumor antigen presentation, recruiting of other immune cells, and directing anti-proliferative and pro-apoptotic effects toward the tumor cells [35,36][20][21]. On the other hand, cancer cells can downregulate or mutate molecules involved in the IFNγ signaling cascade or harness the immunosuppressive functions of IFNγ to evade destructive immunity [34,37][19][22]. Indeed, an increased enrichment of mutated IFNγ signaling related genes such as interferon gamma receptor 1 and 2, Janus kinase 2, and interferon regulatory factor 1 was observed in non-responders to ipilimumab (anti-CTLA-4 mAb) recapitulating the loss of IFNγ signaling-related genes in cancer cells is a mechanism of resistance to anti-CTLA-4 therapy [38][23]. Patients with any of these mutations are likely to be resistant anti-PD-1 therapy due to a lack of PD-L1 expression upon IFNγ exposure [39][24]. MHC-I molecules (also known as human leukocyte antigen or HLA) bind peptides derived from proteins produced in cells and transport them to the cell surface to display antigenic information. It enables CD8+ T cells to identify pathological cells that synthesize abnormal proteins, such as cancer cells that express mutated proteins [40][25]. The absence or acquired poor molecule expression of HLA allows neoantigens to remain undetected by the immune system. Defects in the antigen presentation pathway are caused by a number of mechanisms, including genetic and epigenetic alterations [41][26]. For instance, defects in the MHC-I antigen presentation pathway are the primary mechanism for the loss of tumor-specific MHC-I expression. Downregulated TAP1/TAP2 which results in defective peptide presentation leads to decreased MHC-I stability and its expression on the tumor surface [42][27].
Furthermore, impaired antigen presentation in tumors is caused by structural changes of the MHC-I complex, notably including loss of heterozygosity associated with chromosome 6p21 and from the poor expression of β2M [42][27]. The loss of key transcription factors, such as NF-κB and NLRC5, and epigenetic alterations (such as DNA hypermethylation and downregulation of histone deacetylases) can affect the transcription of MHC-I pathway genes, thereby contributing to immune evasion of cancer cells [42][27]. Downregulation of the NLRC5 transcription factor is associated with decreased expression of target genes such as MHC-I, ß2M, TAP, and immunoproteasome subunits in many cancers, including prostate, lung, uterine, melanoma, and thyroid [43][28]. Interferons are another type of signaling molecule that can induce MHC-I expression [44][29]. The activation of signal transducer and transcription proteins (STAT1, 2, 3) due to type I and type II interferon signaling upregulates MHC-I expression [44][29]. As the function of IFNs in antigen processing and presentation pathway is indispensable, the impairment of IFN signaling leads to the downregulation of MHC-I, implying that this molecular crosstalk has a significant impact on tumorigenesis. Additionally, upregulation of microRNAs (miRNA) inhibits MHC-I expression in several types of cancer including melanoma, esophageal carcinoma, and colorectal cancer. These non-coding RNAs are also found to regulate multiple signaling molecules of antigen presentation pathway, such as TAP1/TAP2 and calreticulin [42][27]. MHC-I expression abnormalities are classified as irreversible or reversible based on the ability to restore molecule expression with cytokine or pharmaceutical treatment [45][30]. MHC-I downregulation is frequently associated with reduced TILs which may correlate with poor clinical outcomes in many cancers, including melanoma, glioblastoma, colorectal, bladder, uterine, cervical, head/neck, breast, and others [46][31]. As a result, the MHC-I downregulation may be used as a prognostic factor for immunotherapy. Thus, understanding pathogenesis by determining whether there are potential ways to restore MHC-I expression can augment the immune system to control malignancies.
The role of epigenetic changes in cancer cells is still debated. On the one hand, these modifications affect antigen processing, presentation, and immune evasion through changes in the expression of immune-related genes, suggesting that using demethylation agents may have therapeutic implications [47,48][32][33]. For example, T helper cells with demethylated DNA could be used as antigen presenting cells (APCs) to generate cytotoxic T lymphocytes (CTLs) and natural killer cells to target cancers [49][34]. On the other hand, the loss of DNA methylation as a result of cell proliferation accompanied by high mutation and copy number load can promote tumor immune evasion [50][35]. TSA, a histone deacetylase inhibitor (HDCAi), promotes the expansion of a population of myeloid-derived suppressor cells (CD11b+Ly6C+F4/80intCD115+), implying that acetylation influences myeloid cell differentiation [51][36]. In contrast, Wang and colleagues reported that using HDACi SAHA reduces MDSC accumulation in the spleen, blood, and tumor bed. Furthermore, exposure of bone marrow cells to SAHA eliminates GM-CSF-induced MDSC population through increased intracellular ROS [52][37].
2.2. Tumor Extrinsic Factors of Resistance to Immunotherapy
The presence of immune cells in human tumors implicates the mixed role of immune cells in tumor growth and progression (Figure 1C). Macrophages, myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), effector T cells (Teffs), DCs, natural killer cells (NKs), and B cells are among the most common immune cells in the TME. Each immune cell subset contributes to pro- or anti-tumor immunity with distinct mechanisms.
Tregs are an important subset of T cells that helps to prevent excessive immune responses and autoimmunity. Tregs can infiltrate human tumors and promote tumor growth [53][38]. These FoxP3-expressing cells either directly impede Teff responses by physical contact, or indirectly by suppressing the latter via the secretion of inhibitory cytokines, including IL-10, IL-35, and TGF-β [54,55,56,57][39][40][41][42]. Upon anti-CTLA-4 mAb treatment, the ratio of Teffs to Tregs was positively associated with treatment response, which is dependent on the presence of Fcγ receptor-expressing macrophages [58[43][44],59], suggesting the use of anti-CTLA-4 antibodies with enhanced FcγR binding profiles to achieve robust anti-tumor responses and improved survival [60][45]. In a clinical trial using ipilimumab to treat patients with advanced melanoma, increased TILs were found to be associated with better outcomes [61][46]. A clinical follow-up study demonstrated that while anti-CTLA-4 immunotherapy promoted intra-tumoral Teff infiltration, it did not cause FoxP3+ T cell depletion in human cancers [62][47]. These pioneering studies on the balance between Teffs and Tregs suggest that an increased number of tumor-infiltrating Teffs, rather than the depletion of Tregs may be used to predict sensitivity to anti-CTLA-4 immunotherapy. If the ratio of these two T cell subsets within the TME in response to immunotherapy does not favor Teffs, resistance is likely to occur throughout the course of treatment.
MDSCs comprise a group of neutrophils and monocytes with potent immunosuppressive properties that may mediate immune responses elicited by T cells, B cells and NK cells [63][48]. Human MDSCs express markers such as CD11b+ and CD33+; however, other types of MDSCs include the presence of HLA-DR, CD33, and CD15 [64][49]. Due to their important roles in promoting angiogenesis, tumor invasion, and metastasis, these cells have emerged as therapeutic targets in cancer [63,65,66,67][48][50][51][52]. A high intra-tumoral number of neutrophils has shown a negative correlation with clinical outcomes in patients with cancer [68][53]. Indeed, Si et al. [69][54] using multidimensional imaging, provided direct evidence that MDSCs inhibit the expression of Teff-secreted Granzyme B and Ki67 (markers for cytotoxicity and proliferation of Teff, respectively). The presence of MDSCs in the TME is closely associated with the efficacy of immunotherapy, as blockade of these cells leads to improved pre-clinical [70][55] and clinical outcomes [71][56]. Kaneda et al. [72][57] suggested that the macrophage PI3Kγ is a critical molecular switch that controls immune suppression by inhibiting NF-κB activation and stimulation of C/EBPβ activation, which are AKT- and mTOR-dependent, to promote immune suppression. More importantly, targeting cancer cells by a combination of selective inactivation of macrophage PI3Kγ and ICIs could overcome cancer resistance to checkpoint blockade therapies. In an ICB resistance setting in mice, to overcome resistance, the molecularly selective pharmacological targeting of the gamma isoform of PI3K in myeloid cells restored the sensitivity of immune checkpoint blocking antibodies [73][58].
Tumor associated macrophages (TAMs) are also associated with patient responses to immunotherapy [74[59][60],75], with the M1 subtype promoting anti-tumor immunity and the M2 subtype promoting tumorigenesis. These cells are categorized according to their distinct activation pathways and expression of surface molecule. Recruitment of TAMs to tumor sites is mediated by tumor-derived effector proteins such as CSF-1, VEGF, and chemokines [76][61]. CSF1/CSF1R axis is crucial for the recruitment of TAMs; therefore, targeting CSF1/CSF1R signaling may have synergistic effects with immunotherapy in suppressing refractory tumors [77,78,79,80][62][63][64][65]. A higher density of TAMs is correlated with poor clinical prognosis in cancer patients [81,82][66][67]. In a mouse model of lung adenocarcinoma, Fritz et al. [83][68] found that the depletion of TAMs may reduce tumor growth due to the downregulation of M2 macrophages. It has been proposed that the inactivation of CCL2 and/or CCR2 signaling is attributed to this phenomenon. In the same vein, similar findings were obtained in other cancer types such as T cell lymphoma [84][69], colon cancer [85][70], lung cancer, and breast cancer [85[70][71][72],86,87], suggesting that the use of pharmacological or biological strategies to inhibit or eliminate these macrophages in the TME may improve patients’ outcome.
Cancer causes chronic inflammation, which depending on the context, may support tumor development [88][73]. Hence, anti-tumor responses and signals may also upregulate inhibitory genes and signaling pathways such as IFNγ, CTLA-4, and PD-L1 in immune cells. Activation of T cell via TCR signaling and CD28 co-stimulation increases CTLA-4 expression [6][4]. Increased IFNγ production by Teffs results in increased production of PD-L1 in both cancer cells and immune cells to hinder anti-tumor responses [2]. The production of several immunosuppressive molecules, including indolaimine-2, 3-deoxygenase, and CEACAM1, is induced by the pro-inflammatory IFNγ leading to peripheral tolerance, an inhibition of NK-mediated cytotoxicity and impaired effector T cell function [89,90][74][75]. Both malignant cells and phagocytic cells can produce immunosuppressive cytokines that inhibit local anti-tumor immunity. For example, the immunosuppressive TGF-β stimulates Tregs to regulate angiogenesis and immune responses [91][76]. Moreover, poor prognosis was associated with elevated TGF-β levels in several malignancies [92][77]. Clinical studies have suggested that combining ICI and selective inhibition of TGF-β receptor and that combining radiation therapy with TGF-β inhibition synergistically improve anti-tumor responses [93,94][78][79]. Multiple chemokines and their respective receptors are essential for the migration of immunosuppressive cells including MDSCs and Tregs to tumor sites [95][80]. Tumor cells produce multiple chemokines such as CCL2, CCL5, CCL7, and CXCL8, which bind to MDSCs CCR1, CCR2, or CXCR2 to recruit MDSCs to the TME [96,97][81][82]. Inhibiting these chemokine receptors or targeting them in combination with ICB may reduce immune evasion and promote anti-tumor T cell responses [98,99,100][83][84][85].
Cancer-associated fibroblasts (CAFs) are a diverse stromal cell population with multiple functions such as matrix deposition and remodeling, crosstalk with infiltrating leukocytes and reciprocal interactions with cancer cells [101][86]. The existence and the role of activated CAFs in the microenvironment are linked to a poor prognosis in various cancers [102][87]. CAFs have been implicated in influencing the function of various immune cells toward an immunosuppressive phenotype via multiple mechanisms, in addition to their ability to recruit immune cells that promote tumor growth. Notably, a recent understanding of CAF heterogeneity in origin and function suggests that driving immune suppression may be mediated by distinct subpopulations of CAFs. For example, mesenchymal stromal cells (MSCs) derived from bone marrow are a significant source of CAFs in breast cancer [103][88]. Surprisingly, MSCs also mediate immunosuppression in the physiological wound healing process. Pro-inflammatory cytokines (IFN, TNF, or IL-1) induce MSCs to produce iNOS, suppressing T cell function in acute liver injury models [104][89]. Tumors may alter the physiological functions of MSCs and fibroblasts to induce the formation of an immunosuppressive TME via the effect of CAFs on specific immune cell populations [105][90]. T-cell exclusion plays a vital role in shaping the low response rate to immunotherapy in CAF-rich tumors [106][91]. Different immunotherapy modalities such as anticancer vaccination, and anti-PD-1 allow the heterogeneous cell population to suppress the response by excluding CD8+ T cells but not CD4+ T cells or macrophages from tumors. T-cell exclusion is associated with increased CTLA-4 expression, which can be inhibited to overcome lymphocyte exclusion [106][91]. Thus, identifying and targeting novel resistance mechanisms such as CAF-related signaling would improve cancer therapies. However, CAFs research is particularly challenging to translate into clinical benefit as CAFs can either promote or inhibit tumorigenesis [101][86].
Other more complex factors, such as age, gender, pre-existing conditions, and intestinal flora, may also contribute toward cancer resistance to immunotherapy. Aging is frequently accompanied by deteriorating immunity and various health-related issues, thus there is a perception that the efficacy of anti-cancer therapeutic approaches may also decline with age. However, in a meta-analysis of randomized controlled trials with ICB, age was not determined to be a factor in how patients responded to the therapy [107][92]. Systemic immunity is influenced by sex and gender. Women are known to mount more robust immune responses than men [104][89]. Another meta-analysis of randomized controlled trials of immune checkpoint therapy for melanoma and human non-small cell lung carcinoma (NSCLC) also revealed that the magnitude of benefit is sex-dependent, with women having a lower pooled hazard ratio for overall survival than men [105][90]. There is also a knowledge gap regarding how immunotherapy affects the anti-tumor immune response of cancer patients with pre-existing common health conditions such as diabetes, obesity, and hypertension. The benefits of immunotherapy to cancer patients with obesity are ambiguous [108][93]. Diabetes has negative effects on ICB in metastatic NSCLC [109][94]. Mechanistic studies are needed to shed light on the linkage between metabolic pathologies and immunotherapy efficacy.
Gut microbiota have been extensively studied over the last two decades and the role of microbes residing in the human gut has had an impact far beyond infectious diseases. Changes in the gut microbiome have been linked to various metabolic disorders and cancers [110][95]. Because the adaptive immune system shapes the gut flora and vice versa, immunotherapy-responsive cancer patients may have a different gut microbiome composition than non-responders [111][96]. Two pioneering studies on gut microbiome in cancer settings have laid the foundation for the concept that gut microbiota modulates immunotherapy [112,113][97][98]. In murine models, Vétizou et al. [112][97] found that optimal responses to CTLA-4 blockade rely on Bacteroides spp. Oral supplementation with B. fragilis, or its derived polysaccharides, or using B. fragilis-specific ACT-based approach restored the efficacy of anti-CTLA-4 therapy. Commensal Bifidobacterium in the gut flora may modulate anti-tumor immune responses and facilitate PD-L1 blockade’s efficacy [113][98]. Subsequent studies have further characterized the association between human gut microbiota and the outcome of cancer patients treated with immune checkpoint inhibitors [114,115,116][99][100][101]. Upon PD-1 immunotherapy in melanoma patients, enhanced systemic and anti-tumor immunity was reported in responders with favorable gut microbiome, such as the abundance of bacteria of the Ruminococcaceae family [114][99]. Similarly, a clear linkage was found between commensal microbial composition and clinical response in metastatic melanoma patients treated with anti-PD1 therapy [116][101]. B. longum, Collinsella aerofaciens, and Enterococcus faecium were among the most abundant bacteria present in the gut flora of responding patients. Fecal transplantation from responders to mice increased T cell responses, higher efficacy of ICB, and improved immune-mediated tumor control [116][101].
References
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nature Immunol. 2002, 3, 991–998.
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723.
- Del Poggetto, E.; Ho, I.L.; Balestrieri, C.; Yen, E.Y.; Zhang, S.; Citron, F.; Shah, R.; Corti, D.; Diaferia, G.R.; Li, C.Y.; et al. Epithelial memory of inflammation limits tissue damage while promoting pancreatic tumorigenesis. Science 2021, 373, eabj0486.
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736.
- Iwai, Y.; Terawaki, S.; Honjo, T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int. Immunol. 2005, 17, 133–144.
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155.
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801.
- Robert, C.; Ribas, A.; Hamid, O.; Daud, A.; Wolchok, J.D.; Joshua, A.M.; Hwu, W.J.; Weber, J.S.; Gangadhar, T.C.; Joseph, R.W.; et al. Durable Complete Response After Discontinuation of Pembrolizumab in Patients with Metastatic Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1668–1674.
- Larkin, J.; Lao, C.D.; Urba, W.J.; McDermott, D.F.; Horak, C.; Jiang, J.; Wolchok, J.D. Efficacy and Safety of Nivolumab in Patients with BRAF V600 Mutant and BRAF Wild-Type Advanced Melanoma: A Pooled Analysis of 4 Clinical Trials. JAMA Oncol. 2015, 1, 433–440.
- Yang, L.; Li, A.; Lei, Q.; Zhang, Y. Tumor-intrinsic signaling pathways: Key roles in the regulation of the immunosuppressive tumor microenvironment. J. Hematol. Oncol. 2019, 12, 125.
- Liu, C.; Peng, W.; Xu, C.; Lou, Y.; Zhang, M.; Wargo, J.A.; Chen, J.Q.; Li, H.S.; Watowich, S.S.; Yang, Y.; et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 393–403.
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 1499–1509.
- Hu-Lieskovan, S.; Mok, S.; Homet Moreno, B.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci. Transl. Med. 2015, 7, 279ra241.
- Liu, L.; Mayes, P.A.; Eastman, S.; Shi, H.; Yadavilli, S.; Zhang, T.; Yang, J.; Seestaller-Wehr, L.; Zhang, S.Y.; Hopson, C.; et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1639–1651.
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216.
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469.
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235.
- Luke, J.J.; Bao, R.; Sweis, R.F.; Spranger, S.; Gajewski, T.F. WNT/β-catenin Pathway Activation Correlates with Immune Exclusion across Human Cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 3074–3083.
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421.
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386.
- Dunn, G.P.; Bruce, A.T.; Sheehan, K.C.; Shankaran, V.; Uppaluri, R.; Bui, J.D.; Diamond, M.S.; Koebel, C.M.; Arthur, C.; White, J.M.; et al. A critical function for type I interferons in cancer immunoediting. Nat. Immunol. 2005, 6, 722–729.
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e1512.
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e399.
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201.
- Maeurer, M.J.; Gollin, S.M.; Storkus, W.J.; Swaney, W.; Karbach, J.; Martin, D.; Castelli, C.; Salter, R.; Knuth, A.; Lotze, M.T. Tumor escape from immune recognition: Loss of HLA-A2 melanoma cell surface expression is associated with a complex rearrangement of the short arm of chromosome 6. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1996, 2, 641–652.
- Hazini, A.; Fisher, K.; Seymour, L. Deregulation of HLA-I in cancer and its central importance for immunotherapy. J. Immunother. Cancer 2021, 9, e002899.
- Taylor, B.C.; Balko, J.M. Mechanisms of MHC-I Downregulation and Role in Immunotherapy Response. Front. Immunol. 2022, 13, 844866.
- Yoshihama, S.; Roszik, J.; Downs, I.; Meissner, T.B.; Vijayan, S.; Chapuy, B.; Sidiq, T.; Shipp, M.A.; Lizee, G.A.; Kobayashi, K.S. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 5999–6004.
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers 2020, 12, 1760.
- Garrido, F. MHC/HLA Class I Loss in Cancer Cells. Adv. Exp. Med. Biol. 2019, 1151, 15–78.
- Tanaka, K.; Tsuchikawa, T.; Miyamoto, M.; Maki, T.; Ichinokawa, M.; Kubota, K.C.; Shichinohe, T.; Hirano, S.; Ferrone, S.; Dosaka-Akita, H.; et al. Down-regulation of Human Leukocyte Antigen class I heavy chain in tumors is associated with a poor prognosis in advanced esophageal cancer patients. Int. J. Oncol. 2012, 40, 965–974.
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179.
- Roulois, D.; Yau, H.L.; De Carvalho, D.D. Pharmacological DNA demethylation: Implications for cancer immunotherapy. Oncoimmunology 2016, 5, e1090077.
- Kirkin, A.F.; Dzhandzhugazyan, K.N.; Guldberg, P.; Fang, J.J.; Andersen, R.S.; Dahl, C.; Mortensen, J.; Lundby, T.; Wagner, A.; Law, I.; et al. Adoptive cancer immunotherapy using DNA-demethylated T helper cells as antigen-presenting cells. Nat. Commun. 2018, 9, 785.
- Jung, H.; Kim, H.S.; Kim, J.Y.; Sun, J.M.; Ahn, J.S.; Ahn, M.J.; Park, K.; Esteller, M.; Lee, S.H.; Choi, J.K. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat. Commun. 2019, 10, 4278.
- Rosborough, B.R.; Castellaneta, A.; Natarajan, S.; Thomson, A.W.; Turnquist, H.R. Histone deacetylase inhibition facilitates GM-CSF-mediated expansion of myeloid-derived suppressor cells in vitro and in vivo. J. Leukoc. Biol. 2012, 91, 701–709.
- Wang, H.F.; Ning, F.; Liu, Z.C.; Wu, L.; Li, Z.Q.; Qi, Y.F.; Zhang, G.; Wang, H.S.; Cai, S.H.; Du, J. Histone deacetylase inhibitors deplete myeloid-derived suppressor cells induced by 4T1 mammary tumors in vivo and in vitro. Cancer Immunol. Immunother. CII 2017, 66, 355–366.
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089.
- Sundstedt, A.; O’Neill, E.J.; Nicolson, K.S.; Wraith, D.C. Role for IL-10 in suppression mediated by peptide-induced regulatory T cells in vivo. J. Immunol. 2003, 170, 1240–1248.
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787.
- Oida, T.; Zhang, X.; Goto, M.; Hachimura, S.; Totsuka, M.; Kaminogawa, S.; Weiner, H.L. CD4+CD25- T cells that express latency-associated peptide on the surface suppress CD4+CD45RBhigh-induced colitis by a TGF-beta-dependent mechanism. J. Immunol. 2003, 170, 2516–2522.
- Nishikawa, H.; Koyama, S. Mechanisms of regulatory T cell infiltration in tumors: Implications for innovative immune precision therapies. J. Immunother. Cancer 2021, 9, e002591.
- Quezada, S.A.; Peggs, K.S.; Curran, M.A.; Allison, J.P. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J. Clin. Investig. 2006, 116, 1935–1945.
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710.
- Arce Vargas, F.; Furness, A.J.S.; Litchfield, K.; Joshi, K.; Rosenthal, R.; Ghorani, E.; Solomon, I.; Lesko, M.H.; Ruef, N.; Roddie, C.; et al. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell 2018, 33, 649–663.e644.
- Hamid, O.; Schmidt, H.; Nissan, A.; Ridolfi, L.; Aamdal, S.; Hansson, J.; Guida, M.; Hyams, D.M.; Gómez, H.; Bastholt, L.; et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J. Transl. Med. 2011, 9, 204.
- Sharma, A.; Subudhi, S.K.; Blando, J.; Scutti, J.; Vence, L.; Wargo, J.; Allison, J.P.; Ribas, A.; Sharma, P. Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3(+) Regulatory T Cells (Tregs) in Human Cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 1233–1238.
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498.
- Wesolowski, R.; Markowitz, J.; Carson, W.E., 3rd. Myeloid derived suppressor cells—A new therapeutic target in the treatment of cancer. J. Immunother. Cancer 2013, 1, 10.
- Yang, L.; DeBusk, L.M.; Fukuda, K.; Fingleton, B.; Green-Jarvis, B.; Shyr, Y.; Matrisian, L.M.; Carbone, D.P.; Lin, P.C. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004, 6, 409–421.
- Yang, L.; Huang, J.; Ren, X.; Gorska, A.E.; Chytil, A.; Aakre, M.; Carbone, D.P.; Matrisian, L.M.; Richmond, A.; Lin, P.C.; et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008, 13, 23–35.
- Li, K.; Shi, H.; Zhang, B.; Ou, X.; Ma, Q.; Chen, Y.; Shu, P.; Li, D.; Wang, Y. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct. Target. Ther. 2021, 6, 362.
- Solito, S.; Falisi, E.; Diaz-Montero, C.M.; Doni, A.; Pinton, L.; Rosato, A.; Francescato, S.; Basso, G.; Zanovello, P.; Onicescu, G.; et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood 2011, 118, 2254–2265.
- Si, Y.; Merz, S.F.; Jansen, P.; Wang, B.; Bruderek, K.; Altenhoff, P.; Mattheis, S.; Lang, S.; Gunzer, M.; Klode, J.; et al. Multidimensional imaging provides evidence for down-regulation of T cell effector function by MDSC in human cancer tissue. Sci. Immunol. 2019, 4, eaaw9159.
- Kodumudi, K.N.; Weber, A.; Sarnaik, A.A.; Pilon-Thomas, S. Blockade of myeloid-derived suppressor cells after induction of lymphopenia improves adoptive T cell therapy in a murine model of melanoma. J. Immunol. 2012, 189, 5147–5154.
- Meyer, C.; Cagnon, L.; Costa-Nunes, C.M.; Baumgaertner, P.; Montandon, N.; Leyvraz, L.; Michielin, O.; Romano, E.; Speiser, D.E. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol. Immunother. CII 2014, 63, 247–257.
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442.
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature 2016, 539, 443–447.
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327.
- Xiang, X.; Wang, J.; Lu, D.; Xu, X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 75.
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76.
- Neubert, N.J.; Schmittnaegel, M.; Bordry, N.; Nassiri, S.; Wald, N.; Martignier, C.; Tillé, L.; Homicsko, K.; Damsky, W.; Maby-El Hajjami, H.; et al. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci. Transl. Med. 2018, 10, eaan3311.
- Mok, S.; Koya, R.C.; Tsui, C.; Xu, J.; Robert, L.; Wu, L.; Graeber, T.; West, B.L.; Bollag, G.; Ribas, A. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014, 74, 153–161.
- Zhu, Y.; Yang, J.; Xu, D.; Gao, X.M.; Zhang, Z.; Hsu, J.L.; Li, C.W.; Lim, S.O.; Sheng, Y.Y.; Zhang, Y.; et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut 2019, 68, 1653–1666.
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069.
- Hu, W.; Li, X.; Zhang, C.; Yang, Y.; Jiang, J.; Wu, C. Tumor-associated macrophages in cancers. Clin. Transl. Oncol. 2016, 18, 251–258.
- Jung, K.Y.; Cho, S.W.; Kim, Y.A.; Kim, D.; Oh, B.C.; Park, D.J.; Park, Y.J. Cancers with Higher Density of Tumor-Associated Macrophages Were Associated with Poor Survival Rates. J. Pathol. Transl. Med. 2015, 49, 318–324.
- Fritz, J.M.; Tennis, M.A.; Orlicky, D.J.; Lin, H.; Ju, C.; Redente, E.F.; Choo, K.S.; Staab, T.A.; Bouchard, R.J.; Merrick, D.T.; et al. Depletion of tumor-associated macrophages slows the growth of chemically induced mouse lung adenocarcinomas. Front. Immunol. 2014, 5, 587.
- Wu, X.; Schulte, B.C.; Zhou, Y.; Haribhai, D.; Mackinnon, A.C.; Plaza, J.A.; Williams, C.B.; Hwang, S.T. Depletion of M2-like tumor-associated macrophages delays cutaneous T-cell lymphoma development in vivo. J. Investig. Dermatol. 2014, 134, 2814–2822.
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859.
- Luo, Y.; Zhou, H.; Krueger, J.; Kaplan, C.; Lee, S.H.; Dolman, C.; Markowitz, D.; Wu, W.; Liu, C.; Reisfeld, R.A.; et al. Targeting tumor-associated macrophages as a novel strategy against breast cancer. J. Clin. Investig. 2006, 116, 2132–2141.
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637.
- Shaked, Y. The pro-tumorigenic host response to cancer therapies. Nat. Rev. Cancer 2019, 19, 667–685.
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022.
- Gray-Owen, S.D.; Blumberg, R.S. CEACAM1: Contact-dependent control of immunity. Nat. Rev. Immunol. 2006, 6, 433–446.
- Seoane, J.; Gomis, R.R. TGF-β Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb. Perspect. Biol. 2017, 9, a022277.
- Massagué, J. TGFbeta in Cancer. Cell 2008, 134, 215–230.
- Hanks, B.A.; Holtzhausen, A.; Evans, K.; Heid, M.; Blobe, G.C. Combinatorial TGF-β signaling blockade and anti-CTLA-4 antibody immunotherapy in a murine BRAFV600E-PTEN-/- transgenic model of melanoma. J. Clin. Oncol. 2014, 32, 3011.
- Vanpouille-Box, C.; Diamond, J.M.; Pilones, K.A.; Zavadil, J.; Babb, J.S.; Formenti, S.C.; Barcellos-Hoff, M.H.; Demaria, S. TGFβ Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res. 2015, 75, 2232–2242.
- Kohli, K.; Pillarisetty, V.G.; Kim, T.S. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 2022, 29, 10–21.
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014, 6, 237ra267.
- Yang, H.; Zhang, Q.; Xu, M.; Wang, L.; Chen, X.; Feng, Y.; Li, Y.; Zhang, X.; Cui, W.; Jia, X. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol. Cancer 2020, 19, 41.
- Sugiyama, D.; Nishikawa, H.; Maeda, Y.; Nishioka, M.; Tanemura, A.; Katayama, I.; Ezoe, S.; Kanakura, Y.; Sato, E.; Fukumori, Y.; et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl. Acad. Sci. USA 2013, 110, 17945–17950.
- Chang, D.K.; Sui, J.; Geng, S.; Muvaffak, A.; Bai, M.; Fuhlbrigge, R.C.; Lo, A.; Yammanuru, A.; Hubbard, L.; Sheehan, J.; et al. Humanization of an anti-CCR4 antibody that kills cutaneous T-cell lymphoma cells and abrogates suppression by T-regulatory cells. Mol. Cancer Ther. 2012, 11, 2451–2461.
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 2020, 26, 878–885.
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186.
- Servais, C.; Erez, N. From sentinel cells to inflammatory culprits: Cancer-associated fibroblasts in tumour-related inflammation. J. Pathol. 2013, 229, 198–207.
- Raz, Y.; Cohen, N.; Shani, O.; Bell, R.E.; Novitskiy, S.V.; Abramovitz, L.; Levy, C.; Milyavsky, M.; Leider-Trejo, L.; Moses, H.L.; et al. Bone marrow–derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med. 2018, 215, 3075–3093.
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638.
- Conforti, F.; Pala, L.; Bagnardi, V.; De Pas, T.; Martinetti, M.; Viale, G.; Gelber, R.D.; Goldhirsch, A. Cancer immunotherapy efficacy and patients’ sex: A systematic review and meta-analysis. Lancet Oncol. 2018, 19, 737–746.
- Ford, K.; Hanley, C.J.; Mellone, M.; Szyndralewiez, C.; Heitz, F.; Wiesel, P.; Wood, O.; Machado, M.; Lopez, M.A.; Ganesan, A.P.; et al. NOX4 Inhibition Potentiates Immunotherapy by Overcoming Cancer-Associated Fibroblast-Mediated CD8 T-cell Exclusion from Tumors. Cancer Res. 2020, 80, 1846–1860.
- Nishijima, T.F.; Muss, H.B.; Shachar, S.S.; Moschos, S.J. Comparison of efficacy of immune checkpoint inhibitors (ICIs) between younger and older patients: A systematic review and meta-analysis. Cancer Treat. Rev. 2016, 45, 30–37.
- Murphy, W.J.; Longo, D.L. The Surprisingly Positive Association Between Obesity and Cancer Immunotherapy Efficacy. JAMA 2019, 321, 1247–1248.
- Jacobi, O.; Landman, Y.; Reinhorn, D.; Icht, O.; Sternschuss, M.; Rotem, O.; Finkel, I.; Allen, A.M.; Dudnik, E.; Goldstein, D.A.; et al. The Relationship of Diabetes Mellitus to Efficacy of Immune Checkpoint Inhibitors in Patients with Advanced Non-Small Cell Lung Cancer. Oncology 2021, 99, 555–561.
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725.
- Zhou, C.B.; Zhou, Y.L.; Fang, J.Y. Gut Microbiota in Cancer Immune Response and Immunotherapy. Trends Cancer 2021, 7, 647–660.
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084.
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089.
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103.
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97.
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108.
More