Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jessie Wu and Version 3 by Jessie Wu.
Human papillomaviruses (HPVs) infect the dividing cells of human epithelia and hijack the cellular replication machinery to ensure their own propagation. In the effort to adapt the cell to suit their own reproductive needs, the virus changes a number of processes, amongst which is the ability of the cell to undergo programmed cell death. Viral infections, forced cell divisions and mutations, which accumulate as a result of uncontrolled proliferation, all trigger one of several cell death pathways. THere, we examine the mechanisms employed by HPVs is examined to ensure the survival of infected cells manipulated into cell cycle progression and proliferation.
- E5
- E6
- E7
- HPV
- apoptosis
1. Introduction
Human papillomaviruses (HPVs) are divided into five genera (α, β, γ, µ and ν), based on the open reading frame sequence coding for the L1 capsid protein [1]. The α and β genera are known to cause health problems in humans and, therefore, are the most extensively studied [2][3].
α-HPVs infect the basal layer of actively dividing mucosal epithelia, which can be found surrounding various cavities of the human body. Based on their ability to cause cancer, α-HPVs are classified either as high-risk (HR), which have oncogenic potential, or as low-risk (LR), which mostly cause self-limiting benign warts [4]. Infection by HR HPVs can lead to the malignant transformation of infected cells during prolonged infection, while LR HPVs, such as HPV-6 and -11, cannot lead to this [5]. The International Agency for Research on Cancer (IARC) classified 14 HPV types as class 1 carcinogens, the most prevalent being HPV-16 and HPV-18, with six other types (HPV-31, -33, -35, -45, -52 and -58) occurring at significant frequencies, depending on the geographical regions [4]. HR HPVs are responsible for 99% of all cervical cancer (CC) cases, as well as 30–90% of other anogenital cancers and 40–60% head-and-neck squamous cell carcinomas (HNSCC) [6][7][8].
HPV replication and virion production depend exclusively on the differentiation processes of keratinocytes, which are the primary target cells of HPVs. The virus intervenes in this process in order to prolong the proliferation of infected cells, and this inevitably triggers the signaling cascade that leads to apoptosis. Likewise, uncontrolled cell division leads to contact inhibition and senescence, i.e., anoikis, while the viral infection itself triggers autophagy. For these reasons, HPVs have evolved various strategies to evade programmed cell death and ensure the continued progression of HPV infection, resulting in the propagation of new virions.
2. Apoptosis
Cells that undergo apoptosis cease to grow and divide. Instead of normal functions they start a series of molecular events that lead to their deaths without the spillage of cellular contents into the surrounding area. Researchers can differentiate between two different apoptosis pathways–the intrinsic, dependent on the release of cytochrome C from the mitochondria, and the extrinsic, which is initiated by the binding of the appropriate ligands to the receptors on the cell surface [9].
The intrinsic or mitochondrial pathway of apoptosis is usually triggered when the cell has suffered damage or when the pro-survival signals from its environment are ablated. In these cases, the mitochondrial permeability transition pore opens and pro-apoptotic proteins leak into the cytoplasm [10]. Cytochrome C then induces conformational changes in the APAF1 protein, which exposes its caspase recruitment domain (CARD domain) and oligomerization domains. This allows APAF1 to oligomerize, forming an apoptosome, which has multiple caspase 9-binding sites (CARD domains) [11]. Binding to procaspase 9 leads to its activation, and activated caspase 9 can then activate procaspase 3 and, with it, the caspase signaling cascade [9].
The external or death receptor pathway of apoptosis is activated when death ligands bind to their appropriate receptors on the cell surface. The most comprehensively characterized death receptors are APO-1/Fas (CD95), TRAIL-R1 and TRAIL-R2, as well as TNFR1, all of which are members of the tumor necrosis factor (TNF) receptor gene superfamily [12]. When their corresponding ligands CD95L, TNFα, lymphotoxin-α and TRAIL bind to the receptors, they initiate receptor trimerization, which brings their death domains into close proximity [13]. This allows the recruitment of adaptor molecules, such as FAS-associated death domain (FADD) or TNF receptor (TNFR)-associated death domain (TRADD) and procaspase-8, resulting in the formation of the death-inducing signaling complex (DISC) [14]. Caspase-8 is activated within the DISC and it is able to activate downstream caspases and initiate the caspase cascade [15]. The internal and external pathways of apoptosis are depicted in Figure 1.
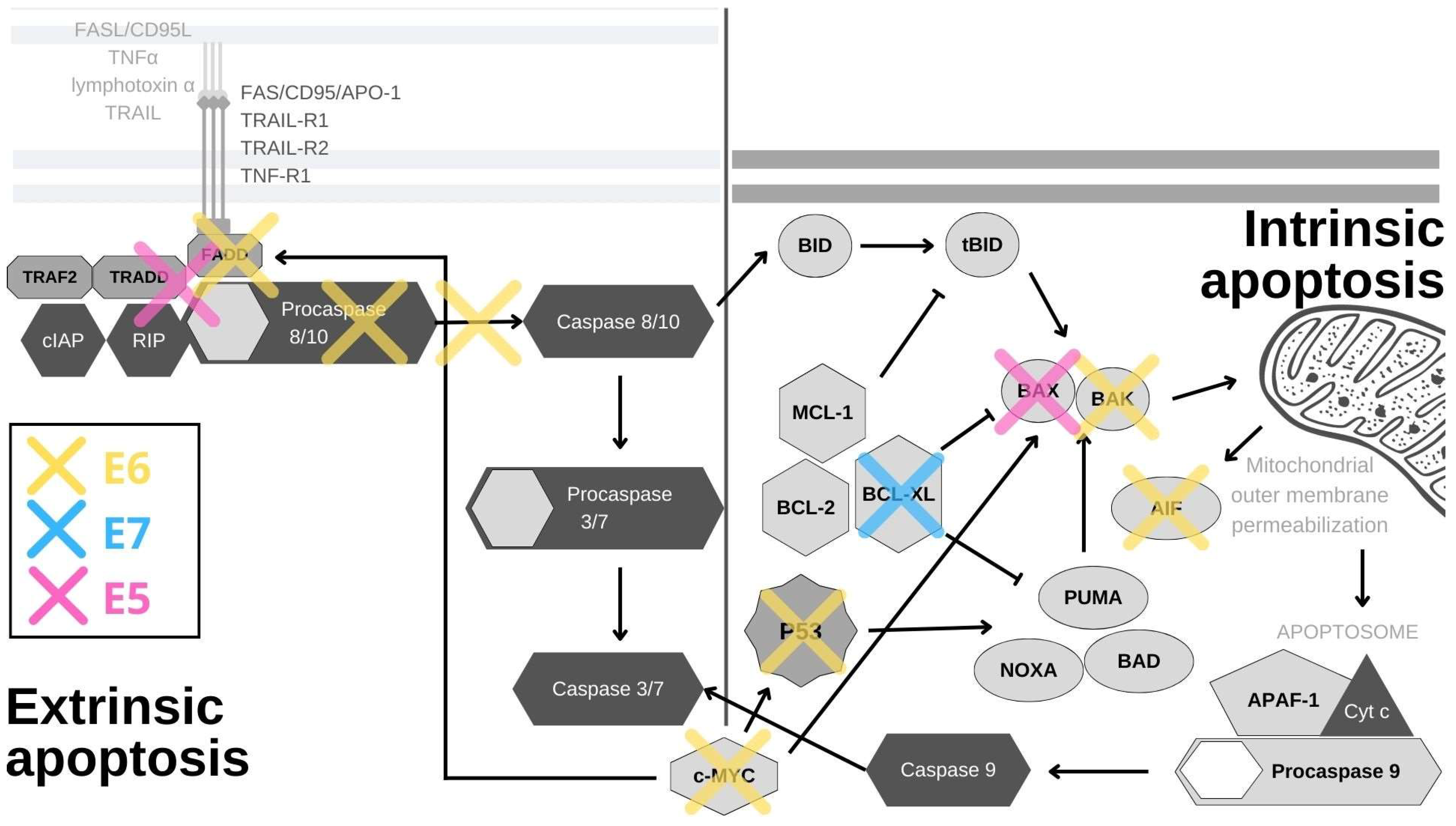
Figure 1. HPV oncoproteins interfere with apoptosis through a variety of mechanisms. E6 (yellow) induces the proteasomal inactivation of p53, the internal detector of cellular stress, and by this activity, it impacts the expression of survivin, YY1 and Cdc2. E6 also degrades the pro-apoptotic proteins BAK, AIF and c-MYC, and cooperates with E5 in inhibiting FAS-mediated apoptosis. By inhibiting TNF-triggered apoptosis, E6 effectively abrogates the external apoptotic pathway and by blocking procaspase activation, it stops the downstream signaling. E6 also utilizes its PDZ-binding motif (PBM) to degrade PDZ domain-containing proteins, leading to the disruption of the apoptotic cascade. E7 (blue) abrogates the binding of SIVA-1 to BCL-XL and thereby prevents the neutralization of anti-apoptotic effects of BCL-XL. E5 (pink) impacts the internal and external apoptotic pathways by inducing the degradation of the pro-apoptotic protein BAK, and impairing the signaling from FAS- and TRAIL-ligands.
A viral infection can also trigger apoptotic signaling within the cell, which is why various viruses have developed different mechanisms to prevent this, HPV being one of them. The inhibition of apoptosis has some particularly long-term consequences in the case of oncogenic viruses, whose transforming properties are amplified by their modulation of programmed cell death pathways. Interestingly, cancers that arise from these viral actions are more resistant to therapies, and therefore uncovering the precise mechanisms of resistance could open potentially very important avenues for therapeutic actions.
p53, the guardian of the genome, is the principal cellular target of HR HPV E6 oncoproteins (Figure 1). Its role is to respond to cellular stress or DNA damage and trigger cell cycle arrest or apoptosis, which it induces mostly by the transcriptional activation of the pro-apoptotic proteins PUMA and NOXA. These proteins then activate other pro-apoptotic proteins in the Bcl2 family, such as Bax and Bak, to induce mitochondrial instability and caspase activation [16]. In normal cells, p53 expression is kept at low levels by the actions of the E3 ubiquitin ligase MDM2. Upon the activation of the DNA damage response, p53 is phosphorylated, which inhibits its binding to MDM2. This leads to p53 accumulation in cells where it acts as a transcription factor for genes involved in cell cycle arrest or cell death [17]. In HPV-infected cells, E6 forms a ternary complex with p53 and the ubiquitin-ligase E6AP (E6-associated protein), which results in the ubiquitination and subsequent degradation of p53 at the proteasome [18][19]. Interestingly, E6/E6AP complex formation is also crucial for E6 protein stability, as E6 by itself quickly undergoes proteasomal degradation [20][21]. Even though p53 is principally inactivated and degraded by the E6/E6AP complex, its function can also be subdued independently of E6AP in a number of ways [22]. The interaction of E6 and p53 inhibits its binding to DNA, possibly due to the changes in protein conformation [23]. By interacting with p53 and its binding partners BCP/p300, hADA3 and TIP60, E6 abrogates the transactivation of p53-responsive genes [24][25][26]. Furthermore, E6 sequesters p53 in the cytoplasm, where p53 cannot exert its influence [23]. Other, often overlooked, tactics for p53 abrogation are post-translational protein and epigenetic modifications. E6 inhibits the p300-mediated acetylation of both p53 and histones, in this way repressing the activation of p53 target genes, among which is p53 itself [23][24][27]. E6 also blocks p53 phosphorylation, which prevents its binding to the p21 promoter, further restraining its function [28]. Conversely, overexpression of p53 was found to be a predictive marker for cisplatin therapy resistance in cases of cervical cancer and mutations in the p53 gene were shown to be connected to radiotherapy resistance in HNSCC [29][30]. In addition, the presence of p53 in the cell is impacted by the upregulation of the YY1 transcription factor [31]. The overexpression of YY1 increases p53 ubiquitination and degradation, and YY1 is upregulated in cervical cancer tissues, which makes it a possible biomarker and a new drug target [31][32]. While the precise mechanism behind this is unknown, YY1 can protect cells from apoptosis, as its inhibition induces p53 activation and cell death in HPV-18 expressing HeLa cells [31]. Mediated by p53 degradation, E6 affects the expression of survivin—a member of the inhibitors of apoptosis (IAP) gene family. E6 was shown to transactivate the survivin promoter, thereby increasing the cell’s resistance to apoptosis [33]. Accordingly, survivin was found to be upregulated in in cervical lesions and could potentially be used as an early marker of cervical carcinogenesis [34]. Surprisingly, in a rare example of an anti-apoptotic effect, E6-mediated p53 degradation leads to the upregulation of Cdc2 and the sensitization of HPV E6-expressing keratinocytes to apoptosis in response to therapeutic agents [35].
In a manner similar to its effect on p53, E6 stimulates E6AP-mediated degradation of BAK and c-MYC proteins (Figure 1). BAK is a pro-apoptotic protein which activates the intrinsic apoptotic program, while c-MYC can trigger the extrinsic and amplify the intrinsic apoptotic pathways [3][36][37]. Interestingly, the c-MYC gene is found in the chromosomal region most commonly impacted by HPV genome integration (8q24), and c-myc overexpression was found to correlate with HPV amplification, making it a potential biomarker for cervical cancer [38][39] Another E6 substrate is the apoptosis-inducing factor (AIF), a pro-apoptotic flavoprotein involved in the mitochondrial apoptotic pathway, which E6 also binds and targets for degradation [40]. E6 also abrogates the extrinsic apoptotic pathway by subverting FADD signaling. By binding to the death effector domains of the FADD receptor, E6 accelerates its degradation and inhibits FAS- and TRAIL-mediated apoptosis (Figure 1). There is also evidence that this pathway is additionally inhibited in keratinocytes expressing HPV-16 oncoproteins, as a result of the downregulation and cytoplasmic sequestration of the TNF receptor 1, combined with a shift towards the expression of a type 2 TNF receptor, which has a weaker response to TNF-α stimuli [41]. Additionally, E6 was recently found to interact with DAXX, another protein involved in FAS-mediated apoptosis, and, in this manner, to decrease the rate of apoptosis. E6 also inhibits TNF-triggered extrinsic apoptosis by binding TNF R1 [42][43][44][45]. In addition to directly inhibiting cell death, E6 can also activate pro-survival pathways. External stimuli, such as pro-inflammatory cytokines, can activate the signal transducer and activator of transcription 3 (STAT3) transcription factor, which has been found to be essential for the survival of cervical cancer cells. E6 activates STAT3 by inducing the expression of the pro-inflammatory cytokine interleukin-6 via the Rac1-NF-kappaB pathway. STAT3 and NF-kappaB co-activation is a specific marker of HPV-positive HNSCC, while STAT3 mRNA detection can be used for cervical lesion screening with great specificity [46][47]. The activation of STAT3 determines the sensitivity of cervical cancer cells to TRAIL-induced apoptosis and can be abrogated by inhibiting the Janus kinase 2 (JAK2), which phosphorylates STAT3 [48][49].
PDZ (PSD95/Dlg/ZO-1) domains are protein–protein interaction modules, 80–90 amino acids in length, that can be found on a large number of proteins. Some of these PDZ domain-containing proteins are well-described targets of the E6/E6AP complex, while some are degraded by E6 independently of E6AP [50]. At its extreme C-terminus, HR HPV E6 contains a Class I PDZ-binding motif (x-T/S-x-L/V) (PBM) through which it interacts with PDZ domain-containing proteins and modulates their functions [51][52]. One of these proteins is MAGI-1, whose degradation facilitates the disruption of tight junctions. Interestingly, restoring MAGI-1 expression in E6-containing cells results in the induction of apoptosis and repression of cell proliferation [52][53]. E6 also utilizes its PBM in NF-kappaB activation, which leads to the accumulation of cellular inhibitor of apoptosis protein 2 (cIAP-2) and subsequently to cellular resistance to TNF-induced apoptosis [54].
The impact of E6 on cell death can also be exerted through DNA methylation. Death-associated protein kinase 1 (DAPK1) is a component of the endoplasmic reticulum (ER) stress-response pathway and a regulator of apoptosis and autophagy [55]. E6 downregulates its expression by inducing the methylation of its promoter, which could be the possible reason behind its inactivation in cervical cancer cells [56][57]. The precise mechanism for this is unknown, but one possible way could be through the upregulation of DNA methyltransferases (DNMTs) [58]. p53 and SP1 bind the DNMT1 promoter and suppress the transcription of the gene. Thus, when E6 degrades and inactivates p53, it effectively increases the expression of DNMT1 [58]. Nevertheless, DNMT1 was found not to have an impact on DAPK1 promoter methylation in lymphoma cells, but a similar mechanism involving other DNMTs is likely to be involved [59]. DAPK1 promoter hypermethylation was found to be correlated with the increasing severity of neoplasia in cervical biopsies and it could potentially be used as a biomarker [60][61]. Furthermore, E6 can bind to the death effector domain of procaspase-8, inhibit its activation and also induce its degradation (Figure 1). Through acting on both receptors and effectors, E6 decreases the activation of both caspase 3 and 8 [42][44]. The two major HPV oncoproteins, E6 and E7, have also been found to upregulate the UHRF1 protein (ubiquitin-like containing PHD and RING finger domain 1) in cells isolated from the early stages of cervical cancer [62]. This protein has been found to bind and regulate DNA methylation in order to regulate gene expression. UHRF1 upregulation by E6/E7 leads to downregulation of gelsolin and UbcH8, which results in inhibition of cell death [62][63].
The role of E7 in apoptosis resistance is much less clearly defined than that of E6. The main cellular target of E7 is the tumor suppressor retinoblastoma protein (pRb). It is normally bound to E2F and this repression complex blocks G1/S cell cycle progression. E7 hijacks the cullin-2 ubiquitin ligase complex and uses it to induce the proteasomal degradation of pRb. The E2F transcription factor is then released and cell cycle progression is ensured [64]. In a similar manner, E7 also degrades the other two pocket proteins, p107 and p130, leading to E2F4 and E2F5 release, respectively, and in this way also further promotes cell cycle progression [2]. Unregulated cell cycle progression would normally trigger apoptosis through a p53-dependent pathway, but, considering that p53 is degraded or inactivated by E6, apoptosis is prevented in cells expressing both oncogenes [65].
Interestingly, E7 was found to sensitize cells to apoptosis, when the Fas receptor was stimulated [66]. Additionally, keratinocytes expressing E7 were found to be more prone to TNF-α and TRAIL-mediated apoptosis, possibly due to the lack of E6 expression, although this is contested by research showing that E7 expression in normal fibroblasts protects the cells from TNF-α and FAS-induced cell death [67][68]. E7 was also found to inhibit apoptosis by enhancing the degradation of a pro-apoptotic protein, insulin-like growth factor-binding protein-3 (IGFBP-3) [69]. In addition, E7 upregulates the expression of catalase and NF-kappaB, through which it conveys the resistance to H2O2-induced cell death [70]. The expression of the cancerous inhibitor of protein phosphatase 2A (CIP2A), an oncoprotein previously implicated in cell proliferation, senescence and apoptosis resistance, was also found to be upregulated by E7 and this upregulation can be used for screening and diagnosis [71]. Furthermore, E7 associates with the pro-apoptotic protein SIVA-1 and disrupts its binding to BCL-XL. The interaction between SIVA-1 and BCL-XL is important for neutralizing the anti-apoptotic effects of BCL-XL, so E7 effectively decreases the rate of apoptosis by abrogating this interaction (Figure 1) [72]. Among other binding partners, E7 also binds histone deacetylases (HDACs) and this interaction was found to contribute to apoptosis resistance, as inhibiting HDACs in E7-expressing cells sensitizes cells to apoptosis and even causes apoptosis by disrupting the mitochondrial transmembrane potential [73][74][75].
E5 has also been implicated in apoptosis modulation and, similarly to E6, it has been found to induce degradation of a pro-apoptotic Bcl-2 family member, in this case Bax, through increased ubiquitination (Figure 1) [76]. During HPV infection, E5 enhances the activation of EGFR, and the PI3K-AKT and ERK1/2 MAPK signaling pathways, all of which have been found to be major survival components [77]. E5 also prevents the external pathway of apoptosis by impairing the formation of DISC via the FAS or TRAIL ligands (Figure 1) [78]. E5 also represses the expression of 179 components of the endoplasmic reticulum that are involved in stress-response pathways [79]. Taking all this together, E5 does not seem to have a decisive role in the evasion of apoptosis, but certainly has an impact on it through several different mechanisms. As E5 expression is found only in a small portion of HPV-induced cancers, its significance for apoptosis evasion in malignancies is not yet clear [80][81].
Since apoptosis is the principal method of programed cell death, HPVs have developed a number of different ways to evade it and ensure the survival of infected cells, the most important of which are summarized in Figure 1. The three viral oncoproteins each control different aspects of this process in order to secure the completion of the viral life cycle and the release of viral progeny. Of the three, and based on the sheer number of interacting partners and signaling pathways it impacts, E6 seems to be the most important for subverting apoptosis. In the general scheme of HPV infections and carcinogenesis, E6 ensures the survival of infected cells, while E7 promotes proliferation and E5 is thought to play a supportive role. Nevertheless, in regards to cell death evasion, HPV oncoproteins sometimes overlap in function. Unfortunately, after the integration of the viral genome into the host cell DNA, which leads to the uncontrolled expression of viral E6 and E7 oncoproteins, these evasion strategies become the major drivers of cancer resistance to therapy.
References
- Bzhalava, D.; Eklund, C.; Dillner, J. International Standardization and Classification of Human Papillomavirus Types. Virology 2015, 476, 341–344.
- Tomaić, V. Functional Roles of E6 and E7 Oncoproteins in HPV-Induced Malignancies at Diverse Anatomical Sites. Cancers 2016, 8, 95.
- Đukić, A.; Lulić, L.; Thomas, M.; Skelin, J.; Saidu, N.E.B.; Grce, M.; Banks, L.; Tomaić, V. HPV Oncoproteins and the Ubiquitin Proteasome System: A Signature of Malignancy? Pathogens 2020, 9, 133.
- World Health Organisation International Agency for Research on Cancer Iarc Monographs on the Evaluation of Carcinogenic Risks to Humans Volume 90 Human Papillomaviruses; International Agency for Research on Cancer: Lyon Cedex, France, 2007; Volume 90, ISBN 9789283212904.
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A Review of Human Carcinogens--Part B: Biological Agents. Lancet Oncol. 2009, 10, 321–322.
- Lont, A.P.; Kroon, B.K.; Horenblas, S.; Gallee, M.P.W.; Berkhof, J.; Meijer, C.J.L.M.; Snijders, P.J.F. Presence of High-Risk Human Papillomavirus DNA in Penile Carcinoma Predicts Favorable Outcome in Survival. Int. J. Cancer 2006, 119, 1078–1081.
- Madsen, B.S.; Jensen, H.L.; Van Den Brule, A.J.C.; Wohlfahrt, J.; Frisch, M. Risk Factors for Invasive Squamous Cell Carcinoma of the Vulva and Vagina-Population-Based Case-Control Study in Denmark. Int. J. Cancer 2008, 122, 2827–2834.
- Li, N.; Franceschi, S.; Howell-Jones, R.; Snijders, P.J.F.; Clifford, G.M. Human Papillomavirus Type Distribution in 30,848 Invasive Cervical Cancers Worldwide: Variation by Geographical Region, Histological Type and Year of Publication. Int. J. Cancer 2011, 128, 927–935.
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592.
- Xiong, S.; Mu, T.; Wang, G.; Jiang, X. Mitochondria-Mediated Apoptosis in Mammals. Protein Cell 2014, 5, 737–749.
- Dorstyn, L.; Akey, C.W.; Kumar, S. New Insights into Apoptosome Structure and Function. Cell Death Differ. 2018, 25, 1194–1208.
- Walczak, H. Death Receptor-Ligand Systems in Cancer, Cell Death, and Inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008698.
- Gough, P.; Myles, I.A. Tumor Necrosis Factor Receptors: Pleiotropic Signaling Complexes and Their Differential Effects. Front. Immunol. 2020, 11, 585880.
- Sessler, T.; Healy, S.; Samali, A.; Szegezdi, E. Structural Determinants of DISC Function: New Insights into Death Receptor-Mediated Apoptosis Signalling. Pharmacol. Ther. 2013, 140, 186–199.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541.
- Roufayel, R.; Younes, K.; Al-Sabi, A.; Murshid, N. BH3-Only Proteins Noxa and Puma Are Key Regulators of Induced Apoptosis. Life 2022, 12, 256.
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The Multiple Mechanisms That Regulate P53 Activity and Cell Fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210.
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. Localization of the E6-AP Regions That Direct Human Papillomavirus E6 Binding, Association with P53, and Ubiquitination of Associated Proteins. Mol. Cell. Biol. 1993, 13, 4918–4927.
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP Complex Functions as a Ubiquitin-Protein Ligase in the Ubiquitination of P53. Cell 1993, 75, 495–505.
- Tomaić, V.; Pim, D.; Banks, L. The Stability of the Human Papillomavirus E6 Oncoprotein Is E6AP Dependent. Virology 2009, 393, 7–10.
- Kranjec, C.; Tomaić, V.; Massimi, P.; Nicolaides, L.; Doorbar, J.; Banks, L. The High-Risk HPV E6 Target Scribble (HScrib) Is Required for HPV E6 Expression in Cervical Tumour-Derived Cell Lines. Papillomavirus Res. 2016, 2, 70–77.
- Massimi, P.; Shai, A.; Lambert, P.; Banks, L. HPV E6 Degradation of P53 and PDZ Containing Substrates in an E6AP Null Background. Oncogene 2008, 27, 1800–1804.
- Lechner, M.S.; Laimins, L.A. Inhibition of P53 DNA Binding by Human Papillomavirus E6 Proteins. J. Virol. 1994, 68, 4262–4273.
- Zimmermann, H.; Degenkolbe, R.; Bernard, H.U.; O’Connor, M.J. The Human Papillomavirus Type 16 E6 Oncoprotein Can Down-Regulate P53 Activity by Targeting the Transcriptional Coactivator CBP/P300. J. Virol. 1999, 73, 6209–6219.
- Sekaric, P.; Shamanin, V.A.; Luo, J.; Androphy, E.J. HAda3 Regulates P14ARF-Induced P53 Acetylation and Senescence. Oncogene 2007, 26, 6261–6268.
- Jha, S.; Vande Pol, S.; Banerjee, N.S.; Dutta, A.B.; Chow, L.T.; Dutta, A. Destabilization of TIP60 by Human Papillomavirus E6 Results in Attenuation of TIP60-Dependent Transcriptional Regulation and Apoptotic Pathway. Mol. Cell 2010, 38, 700–711.
- Thomas, M.C.; Chiang, C.-M. E6 Oncoprotein Represses P53-Dependent Gene Activation via Inhibition of Protein Acetylation Independently of Inducing P53 Degradation. Mol. Cell 2005, 17, 251–264.
- Ajay, A.K.; Meena, A.S.; Bhat, M.K. Human Papillomavirus 18 E6 Inhibits Phosphorylation of P53 Expressed in HeLa Cells. Cell Biosci. 2012, 2, 2.
- Ji, L.; Gong, C.; Ge, L.; Song, L.; Chen, F.; Jin, C.; Zhu, H.; Zhou, G. Orphan Nuclear Receptor Nurr1 as a Potential Novel Marker for Progression in Human Pancreatic Ductal Adenocarcinoma. Exp. Ther. Med. 2017, 13, 551–559.
- Basheeth, N.; Patil, N. Biomarkers in Head and Neck Cancer an Update. Indian J. Otolaryngol. Head neck Surg. Off. Publ. Assoc. Otolaryngol. India 2019, 71, 1002–1011.
- He, G.; Wang, Q.; Zhou, Y.; Wu, X.; Wang, L.; Duru, N.; Kong, X.; Zhang, P.; Wan, B.; Sui, L.; et al. YY1 Is a Novel Potential Therapeutic Target for the Treatment of HPV Infection-Induced Cervical Cancer by Arsenic Trioxide. Int. J. Gynecol. cancer Off. J. Int. Gynecol. Cancer Soc. 2011, 21, 1097–1104.
- Warowicka, A.; Broniarczyk, J.; Węglewska, M.; Kwaśniewski, W.; Goździcka-Józefiak, A. Dual Role of YY1 in HPV Life Cycle and Cervical Cancer Development. Int. J. Mol. Sci. 2022, 23, 3453.
- Borbély, Á.A.; Murvai, M.; Kónya, J.; Beck, Z.; Gergely, L.; Li, F.; Veress, G. Effects of Human Papillomavirus Type 16 Oncoproteins on Survivin Gene Expression. J. Gen. Virol. 2006, 87, 287–294.
- Branca, M.; Giorgi, C.; Santini, D.; Di Bonito, L.; Ciotti, M.; Costa, S.; Benedetto, A.; Casolati, E.A.; Favalli, C.; Paba, P.; et al. Survivin as a Marker of Cervical Intraepithelial Neoplasia and High-Risk Human Papillomavirus and a Predictor of Virus Clearance and Prognosis in Cervical Cancer. Am. J. Clin. Pathol. 2005, 124, 113–121.
- Liu, X.; Roberts, J.; Dakic, A.; Zhang, Y.; Schlegel, R. HPV E7 Contributes to the Telomerase Activity of Immortalized and Tumorigenic Cells and Augments E6-Induced HTERT Promoter Function. Virology 2008, 375, 611–623.
- Gross-Mesilaty, S.; Reinstein, E.; Bercovich, B.; Tobias, K.E.; Schwartz, A.L.; Kahana, C.; Ciechanover, A. Basal and Human Papillomavirus E6 Oncoprotein-Induced Degradation of Myc Proteins by the Ubiquitin Pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 8058–8063.
- Thomas, M.; Banks, L. Inhibition of Bak-Induced Apoptosis by HPV-18 E6. Oncogene 1998, 17, 2943–2954.
- Kübler, K.; Heinenberg, S.; Rudlowski, C.; Keyver-Paik, M.-D.; Abramian, A.; Merkelbach-Bruse, S.; Büttner, R.; Kuhn, W.; Schildhaus, H.-U. C-Myc Copy Number Gain Is a Powerful Prognosticator of Disease Outcome in Cervical Dysplasia. Oncotarget 2015, 6, 825–835.
- Ji, W.; Lou, W.; Hong, Z.; Qiu, L.; Di, W. Genomic Amplification of HPV, H-TERC and C-MYC in Liquid-based Cytological Specimens for Screening of Cervical Intraepithelial Neoplasia and Cancer. Oncol Lett 2019, 17, 2099–2106.
- Shimada, M.; Yamashita, A.; Saito, M.; Ichino, M.; Kinjo, T.; Mizuki, N.; Klinman, D.M.; Okuda, K. The Human Papillomavirus E6 Protein Targets Apoptosis-Inducing Factor (AIF) for Degradation. Sci. Rep. 2020, 10, 14195.
- Cabeça, T.K.; de Mello Abreu, A.; Andrette, R.; de Souza Lino, V.; Morale, M.G.; Aguayo, F.; Termini, L.; Villa, L.L.; Lepique, A.P.; Boccardo, E. HPV-Mediated Resistance to TNF and TRAIL Is Characterized by Global Alterations in Apoptosis Regulatory Factors, Dysregulation of Death Receptors, and Induction of ROS/RNS. Int. J. Mol. Sci. 2019, 20, 198.
- Filippova, M.; Song, H.; Connolly, J.L.; Dermody, T.S.; Duerksen-Hughes, P.J. The Human Papillomavirus 16 E6 Protein Binds to Tumor Necrosis Factor (TNF) R1 and Protects Cells from TNF-Induced Apoptosis. J. Biol. Chem. 2002, 277, 21730–21739.
- Filippova, M.; Parkhurst, L.; Duerksen-Hughes, P.J. The Human Papillomavirus 16 E6 Protein Binds to Fas-Associated Death Domain and Protects Cells from Fas-Triggered Apoptosis. J. Biol. Chem. 2004, 279, 25729–25744.
- Garnett, T.O.; Filippova, M.; Duerksen-Hughes, P.J. Accelerated Degradation of FADD and Procaspase 8 in Cells Expressing Human Papilloma Virus 16 E6 Impairs TRAIL-Mediated Apoptosis. Cell Death Differ. 2006, 13, 1915–1926.
- Tang, S.; Ding, S.; Yu, L.; Shen, H.; Wan, Y.; Wu, Y. Effects of HPV16 E6 Protein on Daxx-Induced Apoptosis in C33A Cells. Cell. Mol. Biol. Lett. 2020, 25, 38.
- Gaykalova, D.A.; Manola, J.B.; Ozawa, H.; Zizkova, V.; Morton, K.; Bishop, J.A.; Sharma, R.; Zhang, C.; Michailidi, C.; Considine, M.; et al. NF-ΚB and Stat3 Transcription Factor Signatures Differentiate HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinoma. Int. J. Cancer 2015, 137, 1879–1889.
- Fan, Y.; Shen, Z. The Clinical Value of HPV E6/E7 and STAT3 MRNA Detection in Cervical Cancer Screening. Pathol.-Res. Pract. 2018, 214, 767–775.
- Morgan, E.L.; Macdonald, A. Autocrine STAT3 Activation in HPV Positive Cervical Cancer through a Virus-Driven Rac1—NFκB—IL-6 Signalling Axis. PLoS Pathog. 2019, 15, e1007835.
- Morgan, E.L.; Wasson, C.W.; Hanson, L.; Kealy, D.; Pentland, I.; McGuire, V.; Scarpini, C.; Coleman, N.; Arthur, J.S.C.; Parish, J.L.; et al. STAT3 Activation by E6 Is Essential for the Differentiation-Dependent HPV18 Life Cycle. PLoS Pathog. 2018, 14, e1006975.
- Thomas, M.; Narayan, N.; Pim, D.; Tomaić, V.; Massimi, P.; Nagasaka, K.; Kranjec, C.; Gammoh, N.; Banks, L.; Thomas, M.; et al. Human Papillomaviruses, Cervical Cancer and Cell Polarity. Oncogene 2008, 27, 7018–7030.
- Pim, D.; Bergant, M.; Boon, S.S.; Ganti, K.; Kranjec, C.; Massimi, P.; Subbaiah, V.K.; Thomas, M.; Tomaić, V.; Banks, L. Human Papillomaviruses and the Specificity of PDZ Domain Targeting. FEBS J. 2012, 279, 3530–3537.
- Ganti, K.; Broniarczyk, J.; Manoubi, W.; Massimi, P.; Mittal, S.; Pim, D.; Szalmas, A.; Thatte, J.; Thomas, M.; Tomaić, V.; et al. The Human Papillomavirus E6 PDZ Binding Motif: From Life Cycle to Malignancy. Viruses 2015, 7, 3530–3551.
- Kranjec, C.; Massimi, P.; Banks, L. Restoration of MAGI-1 Expression in Human Papillomavirus-Positive Tumor Cells Induces Cell Growth Arrest and Apoptosis. J. Virol. 2014, 88, 7155–7169.
- James, M.A.; Lee, J.H.; Klingelhutz, A.J. Human Papillomavirus Type 16 E6 Activates NF-KappaB, Induces CIAP-2 Expression, and Protects against Apoptosis in a PDZ Binding Motif-Dependent Manner. J. Virol. 2006, 80, 5301–5307.
- Singh, P.; Ravanan, P.; Talwar, P. Death Associated Protein Kinase 1 (DAPK1): A Regulator of Apoptosis and Autophagy. Front. Mol. Neurosci. 2016, 9, 46.
- Banzai, C.; Nishino, K.; Quan, J.; Yoshihara, K.; Sekine, M.; Yahata, T.; Tanaka, K. Promoter Methylation of DAPK1, FHIT, MGMT, and CDKN2A Genes in Cervical Carcinoma. Int. J. Clin. Oncol. 2014, 19, 127–132.
- Yanatatsaneejit, P.; Chalertpet, K.; Sukbhattee, J.; Nuchcharoen, I.; Phumcharoen, P.; Mutirangura, A. Promoter Methylation of Tumor Suppressor Genes Induced by Human Papillomavirus in Cervical Cancer. Oncol. Lett. 2020, 20, 955–961.
- Ekanayake Weeramange, C.; Tang, K.D.; Vasani, S.; Langton-Lockton, J.; Kenny, L.; Punyadeera, C. DNA Methylation Changes in Human Papillomavirus-Driven Head and Neck Cancers. Cells 2020, 9, 1359.
- Frazzi, R.; Cusenza Ylenia, V.; Pistoni, M.; Canovi, L.; Cascione, L.; Bertoni, F.; Merli, F. KLF4, DAPK1 and SPG20 Promoter Methylation Is Not Affected by DNMT1 Silencing and Hypomethylating Drugs in Lymphoma Cells. Oncol Rep 2022, 47, 10.
- Feng, Q.; Balasubramanian, A.; Hawes, S.E.; Toure, P.; Sow, P.S.; Dem, A.; Dembele, B.; Critchlow, C.W.; Xi, L.; Lu, H.; et al. Detection of Hypermethylated Genes in Women with and without Cervical Neoplasia. J. Natl. Cancer Inst. 2005, 97, 273–282.
- Zhang, L.; Tan, W.; Yang, H.; Zhang, S.; Dai, Y. Detection of Host Cell Gene/HPV DNA Methylation Markers: A Promising Triage Approach for Cervical Cancer. Front. Oncol. 2022, 12, 831949.
- Lee, H.J.; Kim, M.J.; Kim, Y.S.; Choi, M.Y.; Cho, G.J.; Choi, W.S. UHRF1 Silences Gelsolin to Inhibit Cell Death in Early Stage Cervical Cancer. Biochem. Biophys. Res. Commun. 2020, 526, 1061–1068.
- Zhang, Q.; Qiao, L.; Wang, X.; Ding, C.; Chen, J.J. UHRF1 Epigenetically Down-Regulates UbcH8 to Inhibit Apoptosis in Cervical Cancer Cells. Cell Cycle 2018, 17, 300–308.
- Huh, K.; Zhou, X.; Hayakawa, H.; Cho, J.-Y.; Libermann, T.A.; Jin, J.; Harper, J.W.; Munger, K. Human Papillomavirus Type 16 E7 Oncoprotein Associates with the Cullin 2 Ubiquitin Ligase Complex, Which Contributes to Degradation of the Retinoblastoma Tumor Suppressor. J. Virol. 2007, 81, 9737–9747.
- Alunni-Fabbroni, M.; Littlewood, T.; Deleu, L.; Caldeira, S.; Giarrè, M.; Dell’ Orco, M.; Tommasino, M. Induction of S Phase and Apoptosis by the Human Papillomavirus Type 16 E7 Protein Are Separable Events in Immortalized Rodent Fibroblasts. Oncogene 2000, 19, 2277–2285.
- Aguilar-Lemarroy, A.; Gariglio, P.; Whitaker, N.J.; Eichhorst, S.T.; Hausen, H.z.; Krammer, P.H.; Rösl, F. Restoration of P53 Expression Sensitizes Human Papillomavirus Type 16 Immortalized Human Keratinocytes to CD95-Mediated Apoptosis. Oncogene 2002, 21, 165–175.
- Basile, J.R.; Zacny, V.; Münger, K. The Cytokines Tumor Necrosis Factor-α (TNF-α) and TNF-Related Apoptosis-Inducing Ligand Differentially Modulate Proliferation and Apoptotic Pathways in Human Keratinocytes Expressing the Human Papillomavirus-16 E7 Oncoprotein. J. Biol. Chem. 2001, 276, 22522–22528.
- Thompson, D.A.; Zacny, V.; Belinsky, G.S.; Classon, M.; Jones, D.L.; Schlegel, R.; Münger, K. The HPV E7 Oncoprotein Inhibits Tumor Necrosis Factor α-Mediated Apoptosis in Normal Human Fibroblasts. Oncogene 2001, 20, 3629–3640.
- Santer, F.R.; Moser, B.; Spoden, G.A.; Jansen-Dürr, P.; Zwerschke, W. Human Papillomavirus Type 16 E7 Oncoprotein Inhibits Apoptosis Mediated by Nuclear Insulin-like Growth Factor-Binding Protein-3 by Enhancing Its Ubiquitin/Proteasome-Dependent Degradation. Carcinogenesis 2007, 28, 2511–2520.
- Shim, J.-H.; Cho, K.-J.; Lee, K.-A.; Kim, S.-H.; Myung, P.-K.; Choe, Y.-K.; Yoon, D.-Y. E7-Expressing HaCaT Keratinocyte Cells Are Resistant to Oxidative Stress-Induced Cell Death via the Induction of Catalase. Proteomics 2005, 5, 2112–2122.
- Zhang, W.; Chen, H.; Chen, Y.; Liu, J.; Wang, X.; Yu, X.; Chen, J.J.; Zhao, W. Cancerous Inhibitor of Protein Phosphatase 2A Contributes to Human Papillomavirus Oncoprotein E7-Induced Cell Proliferation via E2F1. Oncotarget 2015, 6, 5253–5262.
- Severino, A.; Abbruzzese, C.; Manente, L.; Valderas, Á.A.; Mattarocci, S.; Federico, A.; Starace, G.; Chersi, A.; Mileo, A.M.; Paggi, M.G. Human Papillomavirus-16 E7 Interacts with Siva-1 and Modulates Apoptosis in HaCaT Human Immortalized Keratinocytes. J. Cell. Physiol. 2007, 212, 118–125.
- Longworth, M.S.; Laimins, L.A. The Binding of Histone Deacetylases and the Integrity of Zinc Finger-like Motifs of the E7 Protein Are Essential for the Life Cycle of Human Papillomavirus Type 31. J. Virol. 2004, 78, 3533–3541.
- Finzer, P.; Krueger, A.; Stöhr, M.; Brenner, D.; Soto, U.; Kuntzen, C.; Krammer, P.H.; Rösl, F. HDAC Inhibitors Trigger Apoptosis in HPV-Positive Cells by Inducing the E2F-P73 Pathway. Oncogene 2004, 23, 4807–4817.
- Darvas, K.; Rosenberger, S.; Brenner, D.; Fritsch, C.; Gmelin, N.; Krammer, P.H.; Rösl, F. Histone Deacetylase Inhibitor-Induced Sensitization to TNFalpha/TRAIL-Mediated Apoptosis in Cervical Carcinoma Cells Is Dependent on HPV Oncogene Expression. Int. J. Cancer 2010, 127, 1384–1392.
- Oh, J.-M.; Kim, S.-H.; Cho, E.-A.; Song, Y.-S.; Kim, W.-H.; Juhnn, Y.-S. Human Papillomavirus Type 16 E5 Protein Inhibits Hydrogen-Peroxide-Induced Apoptosis by Stimulating Ubiquitin-Proteasome-Mediated Degradation of Bax in Human Cervical Cancer Cells. Carcinogenesis 2010, 31, 402–410.
- Zhang, B.; Spandau, D.F.; Roman, A. E5 Protein of Human Papillomavirus Type 16 Protects Human Foreskin Keratinocytes from UV B-Irradiation-Induced Apoptosis. J. Virol. 2002, 76, 220–231.
- Kabsch, K.; Mossadegh, N.; Kohl, A.; Komposch, G.; Schenkel, J.; Alonso, A.; Tomakidi, P. The HPV-16 E5 Protein Inhibits TRAIL- and FasL-Mediated Apoptosis in Human Keratinocyte Raft Cultures. Intervirology 2004, 47, 48–56.
- Sudarshan, S.R.; Schlegel, R.; Liu, X. The HPV-16 E5 Protein Represses Expression of Stress Pathway Genes XBP-1 and COX-2 in Genital Keratinocytes. Biochem. Biophys. Res. Commun. 2010, 399, 617–622.
- Chang, J.L.; Tsao, Y.P.; Liu, D.W.; Huang, S.J.; Lee, W.H.; Chen, S.L. The Expression of HPV-16 E5 Protein in Squamous Neoplastic Changes in the Uterine Cervix. J. Biomed. Sci. 2001, 8, 206–213.
- Um, S.H.; Mundi, N.; Yoo, J.; Palma, D.A.; Fung, K.; MacNeil, D.; Wehrli, B.; Mymryk, J.S.; Barrett, J.W.; Nichols, A.C. Variable Expression of the Forgotten Oncogene E5 in HPV-Positive Oropharyngeal Cancer. J. Clin. Virol. 2014, 61, 94–100.
More