Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Mingqiu Hu and Version 2 by Vivi Li.
Despite aggressive treatment and androgen-deprivation therapy, most prostate cancer patients ultimately develop castration-resistant prostate cancer (CRPC), which is associated with high mortality rates. However, the mechanisms governing the development of CRPC are poorly understood, and androgen receptor (AR) signaling has been shown to be important in CRPC through AR gene mutations, gene overexpression, co-regulatory factors, AR shear variants, and androgen resynthesis. A growing number of non-AR pathways have also been shown to influence the CRPC progression, including the Wnt and Hh pathways. Moreover, non-coding RNAs have been identified as important regulators of the CRPC pathogenesis.
- destructive resistance prostate cancer
- androgen receptor
- Wnt pathway
- Hh pathway
- ncRNAs
1. Introduction
Prostate cancer is the second leading cause of cancer-related death among men worldwide [1]. In China, relative to the rates from before 1990, PCA incidence has risen by 98.21% while corresponding mortality rates have fallen by 3.82%. Consistently, PCA incidence continues to rise [2]. At the time of initial diagnosis, most of the PCA patients exhibit progressive or metastatic disease in China, with androgen-deprivation therapy (ADT) being the treatment of choice for most of the patients with metastatic PCA (European Society of Urology; Guidelines for the Treatment of Prostate Cancer, 2020) [3]. While initially highly effective, however, the median time that patients respond well to ADT is just 18–24 months, after which the patient will progress to develop castration-resistant prostate cancer (CRPC), which is generally defined based upon serum testosterone levels (<50 ng/dL or 1.7 nmol/L) and biochemical (prostate-specific antigen (PSA) levels increasing three times in a row within one week, with at least two of these increases being by more than 50% of the lowest levels, PSA > 2 ng/mL) or radiological (such as two or more new bone lesions in bone scans or soft tissue lesions based upon solid tumor response assessment criteria) evidence of progression after castration treatment [4].
2. Androgen Receptor Pathways
The androgen receptor (AR) is a 110 kDa 919 amino acid nuclear receptor (NR) family member encoded on the X chromosome (q11-q12) (Figure 1A). The AR is composed of a central DNA-binding domain (DBD), and C-terminal ligand-binding domain (LBD), an N-terminal structural domain (NTD), and a hinge region linking the LBD and DBD [5][6][7][8][5,6,7,8]. The AR is encoded by eight exons (Figure 1), with exon 1 encoding the NTD, exons 2,3 encoding the DBD, and exons 4–8 encoding the LBD [9][10][11][9,10,11].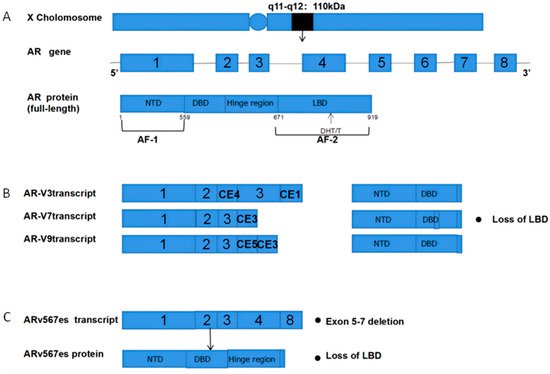
Figure 1. Structural overview of ARs and AR-Vs (AR-V3,7,9 and ARv567es). (A) Structural overview of the AR gene, located on the X chromosome q11-q12, encoding 919 amino acids and consisting of eight exons. The DHT and T ligands to the AR LBD; (B) The mechanisms underlying AR-V3, AR-V7, and AR-V9 production. Exons 4–8 are sheared to produce truncated AR-Vs that lack a LBD and Hinge region; (C) Mechanisms governing the production of ARv567es. Exons 5–7 are missing, resulting in truncated AR proteins and the lack of a LBD. AR = Androgen receptor; AR-Vs = AR variants; NTD = N-terminal transcriptional domain; DBD = DNA-binding domain; LBD = C-terminal ligand-binding domain; CE5 = cryptic exon 5; CE3 = cryptic exon 3; PAS = polyadenylation site; CE4 = cryptic exon.
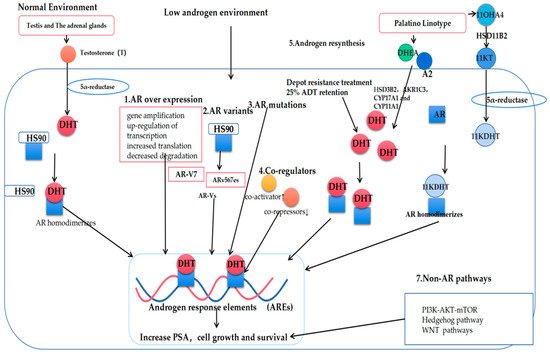
Figure 2. Mechanisms governing the progression of prostate cancer to castration-resistant prostate cancer (CRPC). T: Testosterone; DHT: Dihydrotestosterone; AR: Androgen receptor; AR-Vs: AR variants; DHEA: Dehydroepiandrosterone; A2: androstenedione; 11OXHA4: 11-oxygenated androgens; 11KT: 11-ketotestosterone; 11KDHT: 11-ketodihydrotestosterone.