1. Introduction
The trillions of microbes that inhabit the human gastrointestinal tract have evolved an intimate and mutualistic relationship with their host. The collection of gut microorganisms outnumbers the host in terms of cell numbers as well as genetic composition, with more than 22 million non-redundant genes identified in the gut
[1][2][1,2]—a staggering number when considering that the human genome harbors 20,000–25,000 genes
[3]. Unsurprisingly, this vast and diverse microbial ecosystem plays a vital role in the nutrition and physiology of the host, exerting metabolic functions such as the degradation of complex carbohydrates into short-chain fatty acids (SCFAs), biosynthesis of vitamins, generation of secondary bile acids, and stimulating the development and function of the immune system
[4][5][4,5]. A pivotal component of microbiota–host interactions is the microbial production of a wide array of small-molecule metabolites, which are either synthesized de novo or metabolized from dietary nutrients or host compounds
[6]. These microbiota-derived metabolites act as signaling molecules or metabolic precursors not only in the intestinal milieu, but also in distal organs, following absorption into the circulation
[4][7][4,7].
Diet, geography, ethnicity, age, and genetics dictate the taxonomic profile of the gut microbiome and, consequently, the commensal metabolic capacities
[8][9][10][11][12][13][14][15][8,9,10,11,12,13,14,15]. Perturbations in the composition and metabolic functions of the gut microbiota are associated with a myriad of inflammatory and chronic metabolic diseases, including inflammatory bowel diseases (IBDs), autoimmunity, diabetes, obesity, chronic kidney diseases (CKDs), and cardiovascular diseases (CVDs)
[4][5][7][4,5,7].
In healthy adults, the gut microbiome comprises five main phyla:
Firmicutes,
Bacteroides,
Proteobacteria, Actinobacteria, and
Verrucomicrobia, although
Firmicutes (60–80%) and
Bacteroides (15–25%) are the most abundant
[16]. The introduction of new high-throughput molecular sequencing technologies such as 16S ribosomal RNA (rRNA) gene sequencing and whole-metagenome shotgun sequencing, as well as untargeted metabolomics approaches, has greatly advanced the microbiota research field, and helps in depicting the taxonomic composition and, moreover, the functional genomic and metabolic capacity of commensal bacterial communities. Due to the large interindividual variation in the consortium of microbial species in the human gut, a universal definition of a healthy microbiome remains challenging; nonetheless, microbial diversity with notably high species richness is postulated to be main trait of a healthy microbiome, together with the ability to live in an immunotolerant environment in the host gastrointestinal tract. The high species richness is thought to render the overall microbial community more stable and resilient to environment- and host-derived perturbations, and is partially reflected by the myriad of microbial metabolites that constitute the intestinal milieu and contribute to the pool of circulating metabolites
[6][17][6,17]. Unsurprisingly, studies have shown that disease-associated microbial signatures are correlated with changes in plasma metabolome and microbial metabolite levels. In particular, the imbalance in protective and deleterious microbial metabolites is believed to drive disease progression and severity
[18][19][20][21][22][18,19,20,21,22]. Despite advances in metagenomic and metabolomic approaches as well as culture techniques to characterize the microbiota, only a handful of microbiota-derived byproducts have been identified for their specific roles in health and diseases (
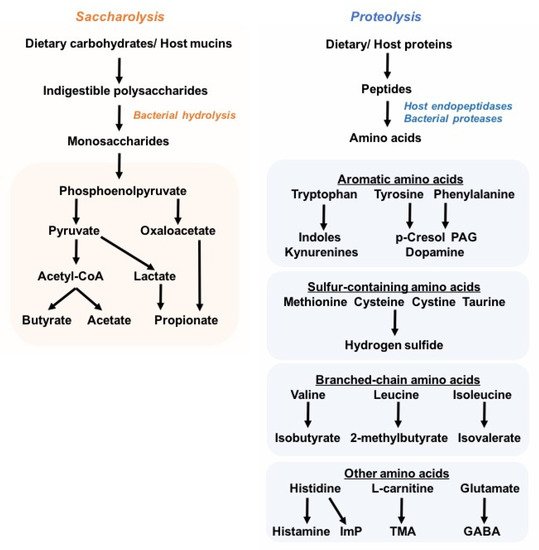
Figure 1). Particularly, the SCFAs butyrate, propionate, and acetate are generated by the bacterial degradation of dietary fibers, and have been extensively studied owing to their health-promoting effects against inflammatory and metabolic disorders, such as colitis and obesity
[23]. In contrast to saccharolytic products, proteolysis-derived microbial metabolites have received less attention, and are largely considered to be deleterious to the host’s (patho)physiology, e.g., trimethylamine N-oxide (TMAO)
[24][25][26][24,25,26], imidazole propionate
[19][27][19,27], phenylacetylglutamine (PAG)
[28][29][30][28,29,30], p-cresol sulfate (PCS), and indoxyl sulfate (IS)
[31][32][33][31,32,33]. Nonetheless, lysine, which is the most abundant amino acid in foods, can be converted into the beneficial butyrate by bacteria related to
Intestinimonas spp.
[34].
2. Tryptophan Catabolism
Although most dietary proteins are absorbed in the small intestine, a relevant proportion of dietary and host proteins (estimated 5–18 g/day) reaches the colon, where proteins are degraded by both host endopeptidases and bacterial proteases
[35][36][37][43,44,45]. The resulting amino acids are further digested and deaminated through bacterial fermentation. Notably, the aromatic amino acids may generate indole and its derivatives, as well as phenolic compounds that may be undesired. Protein fermentation by colonic microbes also contributes to a small proportion of the total microbiota-derived SCFAs (e.g., acetate, propionate, and butyrate), while the branched-chain amino acids give rise to the branched-chain FAs (e.g., isobutyrate, 2-methylbutyrate, isovalerate)
[38][46].
Microbial protein fermentation is favored by low carbohydrate availability, high dietary protein intake, increased colonic pH, and prolonged colonic transit time
[39][40][41][42][47,48,49,50]. In cases of fiber depletion, the bacterial shift from saccharolytic toward proteolytic metabolism results in lower production of beneficial SCFAs; these perturbations in bacterial metabolic activities and the constriction of saccharolytic bacteria are often associated with diseased states and gut-dysbiotic signatures
[4][5][7][18][19][43][44][45][46][47][4,5,7,18,19,51,52,53,54,55]. However, in the case of tryptophan catabolism, the products of its degradation have been linked to both health benefits and poor health outcomes
[7][48][49][50][7,56,57,58].
Tryptophan is one of the nine essential amino acids found in common protein-rich foods, such as milk, cheese, eggs, meat, fish, bananas, oats, nuts, and beans. Tryptophan is largely absorbed in the small intestine, and the fraction that reaches the colon is catabolized by numerous bacterial species, including
Escherichia coli,
Clostridium,
Bacteroides,
Peptostreptococcus,
Eubacterium, and
Lactobacillus species, and
Ruminococcus gnavus [42][51][52][53][54][55][56][57][58][59][50,59,60,61,62,63,64,65,66,67]. In addition, the breakdown of tryptophan by bacteria is not exclusive to the distal colon, as lactobacilli have been reported to catabolize tryptophan in the stomach and small intestine
[48][56].
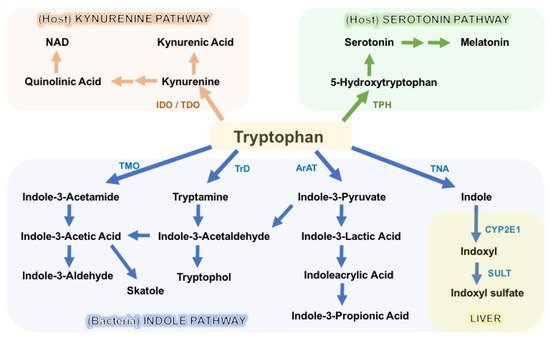
The degradation of tryptophan occurs through three major pathways: the kynurenine pathway (KP), the serotonin pathway, and the indole pathway (
Figure 2). The KP is the primary degradation route in mammalian cells, and encompasses a series of enzymatic steps, with consequential formation of N-formylkynurenine, kynurenine (Kyn), 3-hydroxykynurenine (3-OHKyn), kynurenic acid (kna), 3-hydroxyanthranilic acid (3HAA), quinolinic acid and, ultimately, NAD
+. The rate-limiting step in the KP is catalyzed by indoleamine 2,3-dioxygenase-1/2 (IDO1/2) or tryptophan 2,3-dioxygenase (TDO)
[50][60][58,68]. Following enterocyte-mediated absorption, tryptophan is transported via the hepatic portal system to the liver for utilization by the intrahepatic TDO, whereas the remaining untouched tryptophan is secreted into the circulation for utilization by peripheral tissues. Here, the IDO1 enzyme (expressed in immune and intestinal epithelial cells) is the main driver of tryptophan degradation, as compared to the IDO2 isoform. IDO1 is not constitutively expressed, and it is upregulated in response to inflammation and indigenous gut bacteria. Conversely, IDO1 activity and downstream catabolite formation modulate mucosal reactivity and, hence, microbiota composition
[60][61][68,69]. In addition to host generation of kynurenine, several intestinal bacteria encode homologous KP enzymes, and a few gut microbes (i.e.,
Lactobacillus spp.,
Pseudomonas aeruginosa, and
P. fluorescens) have been shown to produce kynurenine derivatives
[61][62][69,70]. Depending on the targeted tissue and environment, KP metabolites can either exert beneficial effects on host homeostasis or contribute to disease progression. For instance, they promote intestinal homeostasis, immunotolerance, energy expenditure, and resistance to stress-induced depression, but appear deleterious in the context of cancer, metabolic syndrome, and atherosclerosis
[63][64][65][66][67][68][69][71,72,73,74,75,76,77].
Another important catabolite of ingested tryptophan is the neurotransmitter serotonin (5-hydroxytryptamine, 5-HT), which is mainly synthesized by intestinal enterochromaffin cells via tryptophan hydroxylase 1 (TPH1). Serotonin modulates several aspects of host physiology by stimulating (among others) intestinal peristalsis via 5-HT receptor signaling, vasodilatation, and platelet function
[70][78]. A smaller portion of 5-HT is generated in the serotonergic neurons via the THP2 isoform of the enteric and central nervous system, where it modulates mood, appetite, sleep, and cognition
[71][79]. Commensal microbes seem to regulate 5-HT production, since germ-free mice display impaired intestinal production and reduced circulating levels of serotonin
[54][72][62,80]. Although the mechanistic evidence is limited, two studies showed that SCFAs induce the expression of
Tph1, and that the bacterial secondary bile acid deoxycholate can restore the colonic and blood levels of 5-HT
[73][74][81,82]. Moreover, analysis of fecal metagenomes to profile metabolic pathways for neurotransmitter synthesis revealed that gut bacteria have the genomic potential for 5-HT synthesis, although direct evidence of this process is still lacking
[75][83]. Nonetheless, two commensal
Firmicutes bacteria (
Clostridium sporogenes and
Ruminococcus gnavus) possess two phylogenetically distinct enzymes that decarboxylate tryptophan to generate tryptamine—a biogenic amine that stimulates the release of serotonin by enterochromaffin cells
[59][76][67,84]. Moreover, tryptophan decarboxylase homologous genes were found in 9–17% of gut metagenomes of healthy humans—particularly in the genomes of other
Firmicutes, suggesting that more commensal microbes may regulate the release of 5-HT
[59][67].
Lastly, the gut microbes provide a third important route of tryptophan catabolism through the direct transformation of tryptophan into tryptamine and indole metabolites via the action of the bacterial enzymes decarboxylase and tryptophanase A (TnaA), respectively. As described above, bacterial tryptophan decarboxylases have been identified in two gut
Firmicutes:
Clostridium sporogenes and
Ruminococcus gnavus [59][67]. Conversely, the enzyme TnaA is expressed by many Gram-negative and Gram-positive commensal bacteria. Many indigenous microbes have been shown to metabolize tryptophan into indole, including
Escherichia coli,
Clostridium spp.,
Bacteroides spp.,
Lactobacillus spp., and
Streptococcus spp.
[42][48][55][77][78][79][50,56,63,85,86,87]. In addition, gut microbes amplify the variety of tryptophan catabolites through oxidative and reductive pathways generating various indole derivatives. For instance, indole-3-pyruvic acid (IPYA) can be converted into indole-3-lactic acid (ILA), and successively into indole-3-propionic acid (IPA), or IPYA gives rise to indole-3-acetylaldehyde, which is further processed into indole-3-acetic acid (IAA) and, subsequently, into indole-3-aldehyde (IAld)
[61][69] (
Figure 2). Microbiota-produced indoles are detected in the circulation and feces at μM concentrations, and are excreted in the urine
[80][81][82][83][88,89,90,91]. Once absorbed into the circulation, indole can be further converted in the liver into indoxyl sulfate, which has been implicated in the pathogenesis of chronic kidney diseases (CKDs) and cardiovascular comorbidities
[7]. Despite these deleterious effects, most indole derivatives—such as indolelactic acid (ILA), IAA, IPA, and IAld—are key modulators of intestinal homeostasis, endorsing barrier integrity, epithelial renewal, and fine-tuning of mucosal immune responses
[48][84][85][86][56,92,93,94].
In addition, indole and its derivatives act as interspecies signaling molecules in microbial communities by affecting sporulation, drug resistance, and biofilm formation. For instance, ILA has been reported to exert antifungal and antibacterial activities, whereas indole-ethanol (tryptophol) possesses antibacterial and antiphagic properties. Nonetheless, it remains to be elucidated whether these effects substantially modulate the gut microbial ecosystem
[61][87][88][89][69,95,96,97].
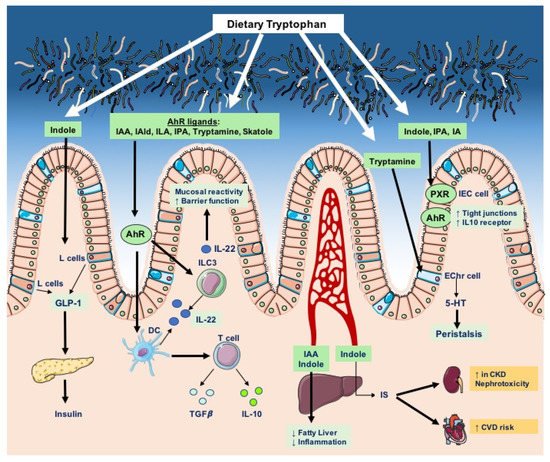
Overall, host and bacterial tryptophan degradation pathways are integral components of host physiology, as they give rise to a variety of bioactive molecules that regulate barrier function, metabolism, inflammation, and endocrine and neuronal activities (Figure 3).
3. Tryptophan Catabolites in Intestinal Homeostasis and Inflammation
Microbial tryptophan catabolites, such as indole and indole derivatives, are potent bioactive molecules that sustain the intestinal barrier’s integrity and contribute to the establishment of immunotolerance against commensal microbes, thereby supporting host–microbiome symbiosis. Bacterial indole derivatives have been largely studied in the context of inflammatory conditions, such as enteric infections or chronic inflammation of the gastrointestinal tract, where their physiological role becomes more manifest.
For instance, the mucin-utilizing bacterium
Peptostreptococcus russellii, which harbors a gene cluster enabling the production of indoleacrylic acid, has been shown to suppress mucosal inflammation and mitigate epithelial injury upon exposure to dextran sodium sulfate (DSS) in mice
[78][86]. Another example of the benefits of microbial tryptophan digestion is given by the commensal bacterium
Lactobacillus reuteri, which was found to expand in conditions of unrestricted tryptophan availability (due to genetic ablation of
Ido1 or administration of tryptophan at high concentrations) in the murine stomach and produce indole-3-aldehyde, resulting in AhR-driven IL-22 expression. This IAld–AhR–IL-22 axis was further shown to provide resistance to fungal infection by
Candida albicans, as well as protection against mucosal inflammation and damage
[48][56]. Furthermore,
L. reuteri can produce indole-3-aldehyde and indole-3-lactic acid, which activate AhR in ILC3 and intraepithelial CD4
+CD8αα
+ double-positive (DP) T cells, respectively
[57][65]. These cells are immunoregulatory T cells that promote oral tolerance and originate from lamina propria CD4 T cells
[90][107]. In mice from different vivaria, the abundance of
L. reuteri is correlated with the number of these intraepithelial DP T cells and, more importantly, colonization with
L. reuteri drives the generation of DP T cells through the generation of indole metabolites that activate AhR in intestinal CD4 T cells
[57][65]. Consistent with a protective effect, another
Lactobacillus sp. (
L. bulgaricus OLL1181) was found to be capable of activating the AhR signaling in intestinal epithelial cells. Indeed, administration of
L. bulgaricus OLL1181 to mice induced the expression of the AhR target gene
Cyp1a1 and ameliorated DSS-induced colitis
[91][125]. Furthermore, administration of three
Lactobacillus strains (
L. murinus CNCM I-5020,
L. reuteri CNCM I-5022, and
L. taiwanensis CNCM I-5019) capable of degrading tryptophan to colitis-susceptible
Card9 (caspase recruitment domain family member 9)-knockout mice attenuates colitis and rescues the mucosal expression of
Il22, and of its target genes
Reg3b and
Reg3g, in an AhR-dependent manner. Importantly, the same study showed that the IBD susceptibility gene
Card9 controls the levels of indigenous microbes and their ability to produce indole derivatives; indeed, the microbiota of
Card9-/- mice displayed a decrease in
Adlercreutzia (genus), Actinobacteria (phylum), and
Lactobacillus reuteri as compared to wild-type mice, as well as impaired production of indole-3-acetic acid. These effects are accompanied by a reduction in intestinal IL-22 expression and the lamina propria ILC3 count. Moreover, transfer of the
Card9-/- microbiota to germ-free mice leads to increased susceptibility to colitis and lower levels of IL-22, indicating that the unbalanced microbiota drives the immune dysregulation
[92][120]. Th
ei
r s study gives an example of the intricate crosstalk between the host and the gut microbiome, and the creation of a vicious cycle in disease where host defects alter the microbiota function which, in turn, contributes to the disease severity.
In addition to the AhR-dependent effects of microbial tryptophan catabolites on gut mucosal integrity, indole-3-propionic acid (IPA) has been shown to fortify barrier integrity via upregulation of tight-junction molecules, by acting as a ligand for the xenobiotic sensor pregnane X receptor (PXR) in IECs
[93][126]. The importance of IPA-mediated gut barrier integrity has further been proven in a subsequent in vivo study employing the IPA-producing gut symbiont
Clostridium sporogenes. Colonization of germ-free mice with wild-type
C. sporogenes resulted in IPA serum concentrations of approximately 80 μM, whereas colonization with a genetically modified form lacking the intact
fldC subunit of the heterotrimeric enzyme phenyllactate dehydratase (necessary for IPA production) resulted in undetectable levels of IPA in serum and the intestinal lumen, and to a marked increase in gut permeability to FITC–dextran as compared to wild-type-colonized mice. This loss of gut barrier function is consistent with global changes in the host immune profile, including increased proportions of neutrophils, classical monocytes, and activated effector/memory CD4 and CD8 T cells
[55][63].
Interestingly, a recent study demonstrated that the microbial indole derivative indole-3-carboxaldehyde modulates the colonic cellular composition during aging, as administration of indole-3-carboxaldehyde to mice promoted intestinal stem cell turnover, enhanced the proportion of goblet cells and, most importantly, rescued the loss of goblet cells in geriatric mice, in an AhR- and IL-10-dependent manner
[94][127]. Strikingly, the protective functions of indoles on the intestinal barrier are seen even in cases of extreme injury caused by irradiation; indeed, indole 3-propionic acid and indole-3-carboxaldehyde have been shown to facilitate gastrointestinal recovery and increase the survival rate in mice following irradiation
[95][96][128,129].
Collectively, most studies support a beneficial and anti-inflammatory function of AhR; however, some pro-inflammatory effects of AhR have been documented. For instance, in Caco-2 intestinal cells, indole has been found to act as an AhR antagonist at concentrations of 100–250 μM
[97][130]. Microbial and dietary oxazoles were shown to induce IDO1 activity and AhR activation but, instead of promoting tolerance, the oxazole–IDO1–AhR axis triggered natural killer T-cell-dependent intestinal inflammation by modulating lipid antigen presentation by IECs and suppressing IL-10 production
[98][131]. In addition, AhR activation has been shown to polarize T cells towards pathogenic Th17 cells in extraintestinal tissues
[99][100][101][101,132,133]. Thus, by sensing exogenous and endogenous molecules, AhR performs multifaceted functions depending on specific ligands and converging signaling from microenvironmental cytokines and factors.
Although the specific roles of many indole derivatives are not yet fully characterized, fine-tuning the microbial tryptophan metabolism holds promise for therapeutic targets in intestinal inflammatory conditions, provided that future investigations can unravel specific gut strains with indole-producing capacity and the bacteria gene clusters necessary for the enzymatic reactions in the generation of indole products.