All processes in human physiology relies on homeostatic mechanisms which require the activation of specific control circuits to adapt the changes imposed by external stimuli. One of the critical modulators of homeostatic balance is autophagy, a catabolic process that is responsible of the destruction of long-lived proteins and organelles through a lysosome degradative pathway. Identification of the mechanism underlying autophagic flux is considered of great importance as both protective and detrimental functions are linked with deregulated autophagy. At the mechanistic and regulatory levels, autophagy is activated in response to diverse stress conditions (food deprivation, hyperthermia and hypoxia), even a novel perspective highlight the potential role of physical forces in autophagy modulation. To understand the crosstalk between all these controlling mechanisms could give uspeople new clues about the specific contribution of autophagy in a wide range of diseases including vascular disorders, inflammation and cancer.
- GPCR
- Gq
- autophagy
- oxidative stress
- mechanotransduction
- extracellular matrix
1. Introduction
2. Autophagy between Nutrient, Mechanical and Oxidative Stress: An Emerging Role of Gαq
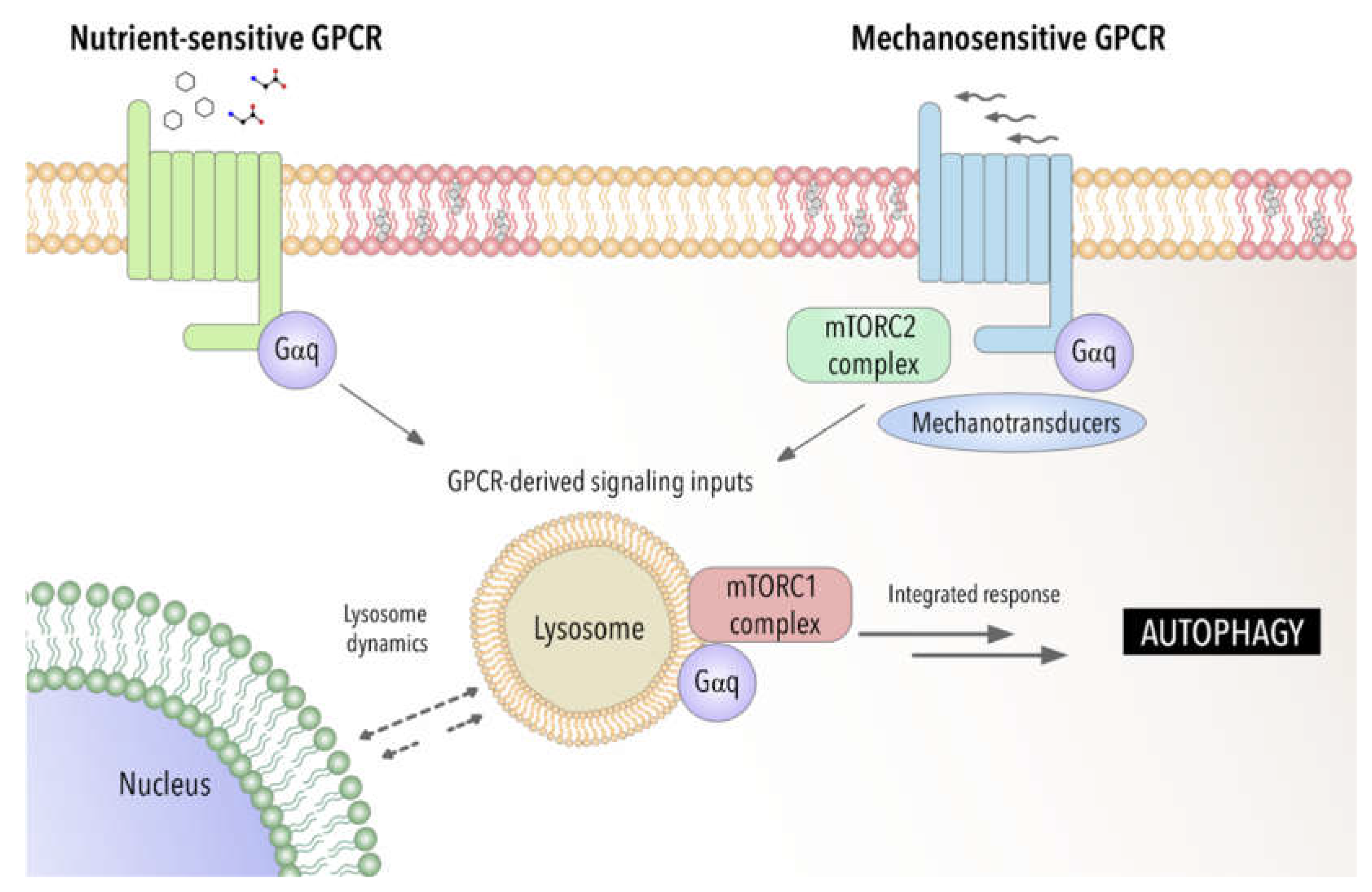
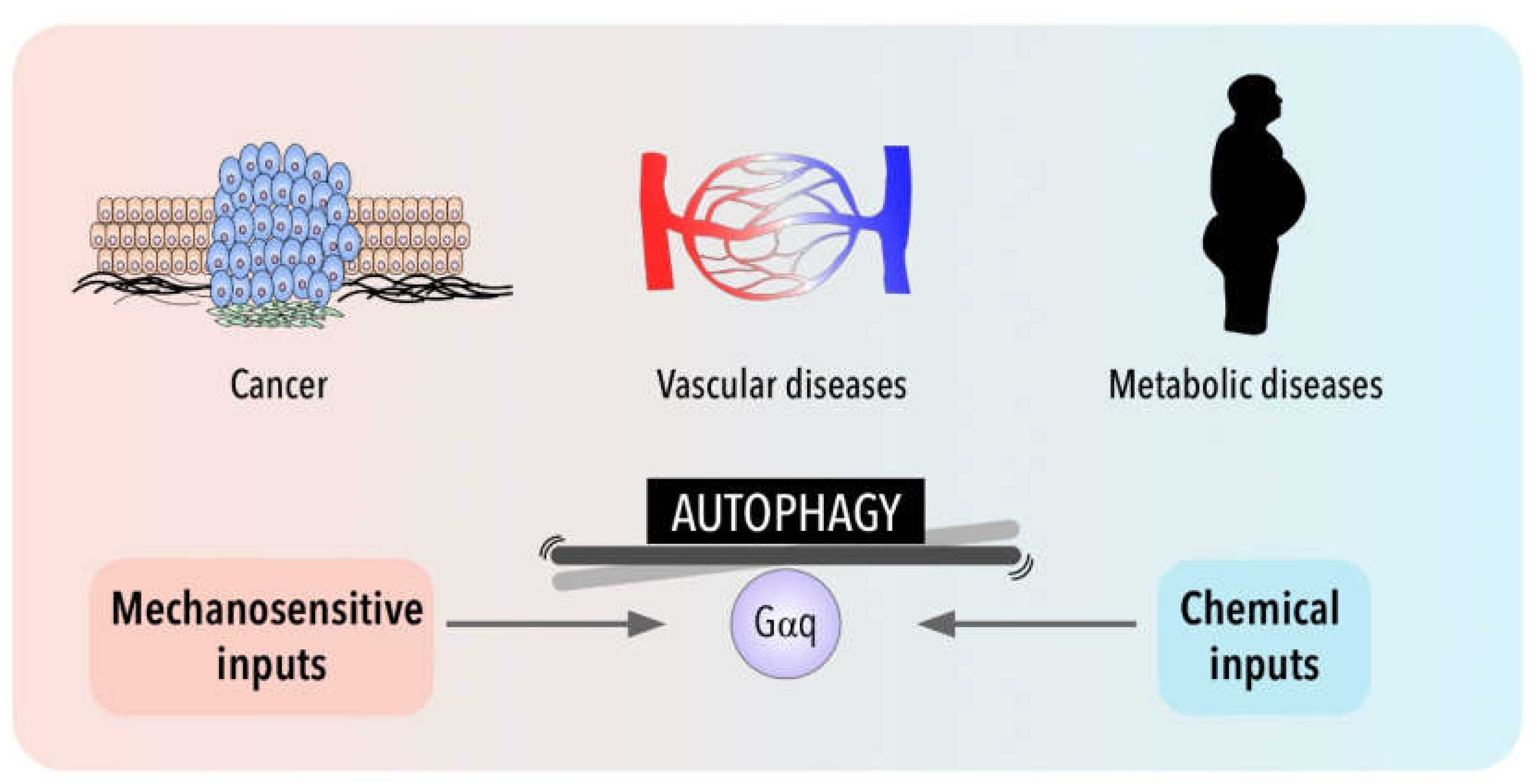
3. Autophagy in Disease for Good and for Bad: Gαq Involvement
References
- Gudipaty, S.A.; Conner, C.M.; Rosenblatt, J.; Montell, D.J. Unconventional Ways to Live and Die: Cell Death and Survival in Development, Homeostasis, and Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 311–332.
- Heldin, C.-H.; Lu, B.; Evans, R.; Gutkind, J.S. Signals and Receptors. Cold Spring Harb. Perspect. Biol. 2016, 8, a005900.
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and Dynamics of GPCR Signaling Complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12.
- Marinissen, M.J.; Gutkind, J.S. G-Protein-Coupled Receptors and Signaling Networks: Emerging Paradigms. Trends Pharmacol. Sci. 2001, 22, 368–376.
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and Extracellular Matrix Homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812.
- Cox, T.R.; Erler, J.T. Remodeling and Homeostasis of the Extracellular Matrix: Implications for Fibrotic Diseases and Cancer. Dis. Model. Mech. 2011, 4, 165–178.
- Mahoney, J.P.; Sunahara, R.K. Mechanistic Insights into GPCR–G Protein Interactions. Curr. Opin. Struct. Biol. 2016, 41, 247–254.
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-Transmembrane Receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650.
- Chen, C.G.; Iozzo, R.V. Extracellular Matrix Guidance of Autophagy: A Mechanism Regulating Cancer Growth. Open Biol. 2022, 12, 210304.
- Hupfer, A.; Brichkina, A.; Koeniger, A.; Keber, C.; Denkert, C.; Pfefferle, P.; Helmprobst, F.; Pagenstecher, A.; Visekruna, A.; Lauth, M. Matrix Stiffness Drives Stromal Autophagy and Promotes Formation of a Protumorigenic Niche. Proc. Natl. Acad. Sci. USA 2021, 118, e2105367118.
- Ge, H.; Tian, M.; Pei, Q.; Tan, F.; Pei, H. Extracellular Matrix Stiffness: New Areas Affecting Cell Metabolism. Front. Oncol. 2021, 11, 631991.
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473.
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy Fights Disease through Cellular Self-Digestion. Nature 2008, 451, 1069–1075.
- Shintani, T.; Klionsky, D.J. Autophagy in Health and Disease: A Double-Edged Sword. Science 2004, 306, 990–995.
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Biol. 2009, 29, 2570–2581.
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular Definitions of Autophagy and Related Processes. EMBO J. 2017, 36, 1811–1836.
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132.
- Dunlop, E.A.; Tee, A.R. MTOR and Autophagy: A Dynamic Relationship Governed by Nutrients and Energy. Semin. Cell Dev. Biol. 2014, 36, 121–129.
- Park, J.-M.; Jung, C.H.; Seo, M.; Otto, N.M.; Grunwald, D.; Kim, K.H.; Moriarity, B.; Kim, Y.-M.; Starker, C.; Nho, R.S.; et al. The ULK1 Complex Mediates MTORC1 Signaling to the Autophagy Initiation Machinery via Binding and Phosphorylating ATG14. Autophagy 2016, 12, 547–564.
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-Dependent MTORC1 Association with the ULK1–Atg13–FIP200 Complex Required for Autophagy. Mol. Biol. Cell 2009, 20, 1981–1991.
- Case, N.; Thomas, J.; Sen, B.; Styner, M.; Xie, Z.; Galior, K.; Rubin, J. Mechanical Regulation of Glycogen Synthase Kinase 3β (GSK3β) in Mesenchymal Stem Cells Is Dependent on Akt Protein Serine 473 Phosphorylation via MTORC2 Protein. J. Biol. Chem. 2011, 286, 39450–39456.
- Hernández-Cáceres, M.P.; Munoz, L.; Pradenas, J.M.; Pena, F.; Lagos, P.; Aceiton, P.; Owen, G.I.; Morselli, E.; Criollo, A.; Ravasio, A.; et al. Mechanobiology of Autophagy: The Unexplored Side of Cancer. Front. Oncol. 2021, 11, 632956.
- Dupont, N.; Codogno, P. Autophagy Transduces Physical Constraints into Biological Responses. Int. J. Biochem. Cell Biol. 2016, 79, 419–426.
- Martinac, B.; Poole, K. Mechanically Activated Ion Channels. Int. J. Biochem. Cell Biol. 2018, 97, 104–107.
- Saotome, K.; Murthy, S.E.; Kefauver, J.M.; Whitwam, T.; Patapoutian, A.; Ward, A.B. Structure of the Mechanically Activated Ion Channel Piezo1. Nature 2018, 554, 481–486.
- Sinha, B.; Köster, D.; Ruez, R.; Gonnord, P.; Bastiani, M.; Abankwa, D.; Stan, R.V.; Butler-Browne, G.; Vedie, B.; Johannes, L.; et al. Cells Respond to Mechanical Stress by Rapid Disassembly of Caveolae. Cell 2011, 144, 402–413.
- Praetorius, H.A. The Primary Cilium as Sensor of Fluid Flow: New Building Blocks to the Model. A Review in the Theme: Cell Signaling: Proteins, Pathways and Mechanisms. Am. J. Physiol. -Cell Physiol. 2015, 308, C198–C208.
- Baschieri, F.; Dayot, S.; Elkhatib, N.; Ly, N.; Capmany, A.; Schauer, K.; Betz, T.; Vignjevic, D.M.; Poincloux, R.; Montagnac, G. Frustrated Endocytosis Controls Contractility-Independent Mechanotransduction at Clathrin-Coated Structures. Nat. Commun. 2018, 9, 3825.
- Kirby, T.J.; Lammerding, J. Emerging Views of the Nucleus as a Cellular Mechanosensor. Nat. Cell Biol. 2018, 20, 373–381.
- Helle, S.C.J.; Feng, Q.; Aebersold, M.J.; Hirt, L.; Grüter, R.R.; Vahid, A.; Sirianni, A.; Mostowy, S.; Snedeker, J.G.; Šarić, A.; et al. Mechanical Force Induces Mitochondrial Fission. eLife 2017, 6, e30292.
- Guet, D.; Mandal, K.; Pinot, M.; Hoffmann, J.; Abidine, Y.; Sigaut, W.; Bardin, S.; Schauer, K.; Goud, B.; Manneville, J.-B. Mechanical Role of Actin Dynamics in the Rheology of the Golgi Complex and in Golgi-Associated Trafficking Events. Curr. Biol. 2014, 24, 1700–1711.
- Aguilera, M.O.; Berón, W.; Colombo, M.I. The Actin Cytoskeleton Participates in the Early Events of Autophagosome Formation upon Starvation Induced Autophagy. Autophagy 2012, 8, 1590–1603.
- Kast, D.J.; Dominguez, R. The Cytoskeleton–Autophagy Connection. Curr. Biol. 2017, 27, R318–R326.
- Hsu, P.; Shi, Y. Regulation of Autophagy by Mitochondrial Phospholipids in Health and Diseases. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2017, 1862, 114–129.
- Avivar-Valderas, A.; Bobrovnikova-Marjon, E.; Alan Diehl, J.; Bardeesy, N.; Debnath, J.; Aguirre-Ghiso, J.A. Regulation of Autophagy during ECM Detachment Is Linked to a Selective Inhibition of MTORC1 by PERK. Oncogene 2013, 32, 4932–4940.
- Cai, Z.; Zhao, D.; Sun, Y.; Gao, D.; Li, X.; Yang, J.; Ma, Z. Detachment-Based Equilibrium of Anoikic Cell Death and Autophagic Cell Survival through Adaptor Protein P66Shc. Anat. Rec. 2016, 299, 325–333.
- Vlahakis, A.; Debnath, J. The Interconnections between Autophagy and Integrin-Mediated Cell Adhesion. J. Mol. Biol. 2017, 429, 515–530.
- Anlaş, A.A.; Nelson, C.M. Soft Microenvironments Induce Chemoresistance by Increasing Autophagy Downstream of Integrin-Linked Kinase. Cancer Res. 2020, 80, 4103–4113.
- Neill, T.; Schaefer, L.; Iozzo, R.V. Instructive Roles of Extracellular Matrix on Autophagy. Am. J. Pathol. 2014, 184, 2146–2153.
- Buraschi, S.; Neill, T.; Goyal, A.; Poluzzi, C.; Smythies, J.; Owens, R.T.; Schaefer, L.; Torres, A.; Iozzo, R.V. Decorin Causes Autophagy in Endothelial Cells via Peg3. Proc. Natl. Acad. Sci. USA 2013, 110, E2582–E2591.
- Nguyen, T.M.B.; Subramanian, I.V.; Xiao, X.; Ghosh, G.; Nguyen, P.; Kelekar, A.; Ramakrishnan, S. Endostatin Induces Autophagy in Endothelial Cells by Modulating Beclin 1 and β-Catenin Levels. J. Cell. Mol. Med. 2009, 13, 3687–3698.
- Castagnaro, S.; Gambarotto, L.; Cescon, M.; Bonaldo, P. Autophagy in the Mesh of Collagen VI. Matrix Biol. 2021, 100–101, 162–172.
- Carmignac, V.; Svensson, M.; Körner, Z.; Elowsson, L.; Matsumura, C.; Gawlik, K.I.; Allamand, V.; Durbeej, M. Autophagy Is Increased in Laminin A2 Chain-Deficient Muscle and Its Inhibition Improves Muscle Morphology in a Mouse Model of MDC1A. Hum. Mol. Genet. 2011, 20, 4891–4902.
- Marullo, S.; Doly, S.; Saha, K.; Enslen, H.; Scott, M.G.H.; Coureuil, M. Mechanical GPCR Activation by Traction Forces Exerted on Receptor N-Glycans. ACS Pharmacol. Transl. Sci. 2020, 3, 171–178.
- Iliff, A.J.; Xu, X.Z.S. A Mechanosensitive GPCR That Detects the Bloody Force. Cell 2018, 173, 542–544.
- Ozkan, A.D.; Gettas, T.; Sogata, A.; Phaychanpheng, W.; Zhou, M.; Lacroix, J.J. Mechanical and Chemical Activation of GPR68 Probed with a Genetically Encoded Fluorescent Reporter. J. Cell Sci. 2021, 134, jcs255455.
- García-Hoz, C.; Sánchez-Fernández, G.; Díaz-Meco, M.T.; Moscat, J.; Mayor, F.; Ribas, C. Gαq Acts as an Adaptor Protein in Protein Kinase Cζ (PKCζ)-Mediated ERK5 Activation by G Protein-Coupled Receptors (GPCR). J. Biol. Chem. 2010, 285, 13480–13489.
- Nigro, P.; Abe, J.; Berk, B.C. Flow Shear Stress and Atherosclerosis: A Matter of Site Specificity. Antioxid. Redox Signal. 2011, 15, 1405–1414.
- Folts, C.J.; Giera, S.; Li, T.; Piao, X. Adhesion G Protein-Coupled Receptors as Drug Targets for Neurological Diseases. Trends Pharmacol. Sci. 2019, 40, 278–293.
- Scholz, N. Cancer Cell Mechanics: Adhesion G Protein-Coupled Receptors in Action? Front. Oncol. 2018, 8, 59.
- Aplin, A.; Jasionowski, T.; Tuttle, D.L.; Lenk, S.E.; Dunn, W.A. Cytoskeletal Elements Are Required for the Formation and Maturation of Autophagic Vacuoles. J. Cell. Physiol. 1992, 152, 458–466.
- Kaushik, S.; Cuervo, A.M. The Coming of Age of Chaperone-Mediated Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381.
- Lim, C.-Y.; Zoncu, R. The Lysosome as a Command-and-Control Center for Cellular Metabolism. J. Cell Biol. 2016, 214, 653–664.
- Cabukusta, B.; Neefjes, J. Mechanisms of Lysosomal Positioning and Movement. Traffic 2018, 19, 761–769.
- Kesisova, I.A.; Robinson, B.P.; Spiliotis, E.T. A Septin GTPase Scaffold of Dynein–Dynactin Motors Triggers Retrograde Lysosome Transport. J. Cell Biol. 2021, 220, e202005219.
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M.; et al. Lysosomal Positioning Coordinates Cellular Nutrient Responses. Nat. Cell Biol. 2011, 13, 453–460.
- Jordens, I.; Fernandez-Borja, M.; Marsman, M.; Dusseljee, S.; Janssen, L.; Calafat, J.; Janssen, H.; Wubbolts, R.; Neefjes, J. The Rab7 Effector Protein RILP Controls Lysosomal Transport by Inducing the Recruitment of Dynein-Dynactin Motors. Curr. Biol. 2001, 11, 1680–1685.
- Bucci, C.; Thomsen, P.; Nicoziani, P.; McCarthy, J.; van Deurs, B. Rab7: A Key to Lysosome Biogenesis. Mol. Biol. Cell 2000, 11, 467–480.
- Xing, R.; Zhou, H.; Jian, Y.; Li, L.; Wang, M.; Liu, N.; Yin, Q.; Liang, Z.; Guo, W.; Yang, C. The Rab7 Effector WDR91 Promotes Autophagy-Lysosome Degradation in Neurons by Regulating Lysosome Fusion. J. Cell Biol. 2021, 220, e202007061.
- Willett, R.; Martina, J.A.; Zewe, J.P.; Wills, R.; Hammond, G.R.V.; Puertollano, R. TFEB Regulates Lysosomal Positioning by Modulating TMEM55B Expression and JIP4 Recruitment to Lysosomes. Nat. Commun. 2017, 8, 1580.
- Settembre, C.; di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433.
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A Lysosome-to-Nucleus Signalling Mechanism Senses and Regulates the Lysosome via MTOR and TFEB. EMBO J. 2012, 31, 1095–1108.
- Cabezudo, S.; Sanz-Flores, M.; Caballero, A.; Tasset, I.; Rebollo, E.; Diaz, A.; Aragay, A.M.; Cuervo, A.M.; Mayor, F.; Ribas, C. Gαq Activation Modulates Autophagy by Promoting MTORC1 Signaling. Nat. Commun. 2021, 12, 4540.
- Wright, S.C.; Lukasheva, V.; le Gouill, C.; Kobayashi, H.; Breton, B.; Mailhot-Larouche, S.; Blondel-Tepaz, É.; Antunes Vieira, N.; Costa-Neto, C.; Héroux, M.; et al. BRET-Based Effector Membrane Translocation Assay Monitors GPCR-Promoted and Endocytosis-Mediated Gq Activation at Early Endosomes. Proc. Natl. Acad. Sci. USA 2021, 118, e2025846118.
- Kenific, C.M.; Wittmann, T.; Debnath, J. Autophagy in Adhesion and Migration. J. Cell Sci. 2016, 129, 3685–3693.
- King, J.S. Mechanical Stress Meets Autophagy: Potential Implications for Physiology and Pathology. Trends Mol. Med. 2012, 18, 583–588.
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in Major Human Diseases. EMBO J. 2021, 40, e108863.
- Ichimura, Y.; Komatsu, M. Pathophysiological Role of Autophagy: Lesson from Autophagy-Deficient Mouse Models. Exp. Anim. 2011, 60, 329–345.
- Kitada, M.; Koya, D. Autophagy in Metabolic Disease and Ageing. Nat. Rev. Endocrinol. 2021, 17, 647–661.
- Rabinowitz, J.D.; White, E. Autophagy and Metabolism. Science 2010, 330, 1344–1348.
- Van Beek, N.; Klionsky, D.J.; Reggiori, F. Genetic Aberrations in Macroautophagy Genes Leading to Diseases. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Res. 2018, 1865, 803–816.
- Zhang, X.; Wu, D.; Wang, C.; Luo, Y.; Ding, X.; Yang, X.; Silva, F.; Arenas, S.; Weaver, J.M.; Mandell, M.; et al. Sustained Activation of Autophagy Suppresses Adipocyte Maturation via a Lipolysis-Dependent Mechanism. Autophagy 2020, 16, 1668–1682.
- Codogno, P.; Meijer, A.J. Autophagy: A Potential Link between Obesity and Insulin Resistance. Cell Metab. 2010, 11, 449–451.
- Madrigal-Matute, J.; Cuervo, A.M. Regulation of Liver Metabolism by Autophagy. Gastroenterology 2016, 150, 328–339.
- Park, E.-Y.; Park, J.-B. High Glucose-Induced Oxidative Stress Promotes Autophagy through Mitochondrial Damage in Rat Notochordal Cells. Int. Orthop. 2013, 37, 2507–2514.
- Oliveira de Souza, C.; Sun, X.; Oh, D. Metabolic Functions of G Protein-Coupled Receptors and β-Arrestin-Mediated Signaling Pathways in the Pathophysiology of Type 2 Diabetes and Obesity. Front. Endocrinol. 2021, 12, 1005.
- Blad, C.C.; Tang, C.; Offermanns, S. G Protein-Coupled Receptors for Energy Metabolites as New Therapeutic Targets. Nat. Rev. Drug Discov. 2012, 11, 603–619.
- Offermanns, S. Free Fatty Acid (FFA) and Hydroxy Carboxylic Acid (HCA) Receptors. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 407–434.
- Wauson, E.M.; Zaganjor, E.; Cobb, M.H. Amino Acid Regulation of Autophagy through the GPCR TAS1R1-TAS1R3. Autophagy 2013, 9, 418–419.
- Wauson, E.M.; Zaganjor, E.; Lee, A.-Y.; Guerra, M.L.; Ghosh, A.B.; Bookout, A.L.; Chambers, C.P.; Jivan, A.; McGlynn, K.; Hutchison, M.R.; et al. The G Protein-Coupled Taste Receptor T1R1/T1R3 Regulates MTORC1 and Autophagy. Mol. Cell 2012, 47, 851–862.
- Wauson, E.M.; Lorente-Rodríguez, A.; Cobb, M.H. Minireview: Nutrient Sensing by G Protein-Coupled Receptors. Mol. Endocrinol. 2013, 27, 1188–1197.
- Lizaso, A.; Tan, K.-T.; Lee, Y.-H. β-Adrenergic Receptor-Stimulated Lipolysis Requires the RAB7-Mediated Autolysosomal Lipid Degradation. Autophagy 2013, 9, 1228–1243.
- Jung, H.S.; Chung, K.W.; Won Kim, J.; Kim, J.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.-H.; Kim, J.-W.; et al. Loss of Autophagy Diminishes Pancreatic β Cell Mass and Function with Resultant Hyperglycemia. Cell Metab. 2008, 8, 318–324.
- Moscat, J.; Diaz-Meco, M.T. Feedback on Fat: P62-MTORC1-Autophagy Connections. Cell 2011, 147, 724–727.
- Onken, M.D.; Noda, S.E.; Kaltenbronn, K.M.; Frankfater, C.; Makepeace, C.M.; Fettig, N.; Piggott, K.D.; Custer, P.L.; Ippolito, J.E.; Blumer, K.J. Oncogenic Gq/11 Signaling Acutely Drives and Chronically Sustains Metabolic Reprogramming in Uveal Melanoma. J. Biol. Chem. 2022, 298, 101495.
- Kimura, T.; Pydi, S.P.; Wang, L.; Haspula, D.; Cui, Y.; Lu, H.; König, G.M.; Kostenis, E.; Steinberg, G.R.; Gavrilova, O.; et al. Adipocyte Gq Signaling Is a Regulator of Glucose and Lipid Homeostasis in Mice. Nat. Commun. 2022, 13, 1652.
- Romani, P.; Valcarcel-Jimenez, L.; Frezza, C.; Dupont, S. Crosstalk between Mechanotransduction and Metabolism. Nat. Rev. Mol. Cell Biol. 2021, 22, 22–38.
- Kim, S.W.; Ehrman, J.; Ahn, M.-R.; Kondo, J.; Lopez, A.A.M.; Oh, Y.S.; Kim, X.H.; Crawley, S.W.; Goldenring, J.R.; Tyska, M.J.; et al. Shear Stress Induces Noncanonical Autophagy in Intestinal Epithelial Monolayers. Mol. Biol. Cell 2017, 28, 3043–3056.
- Miceli, C.; Roccio, F.; Penalva-Mousset, L.; Burtin, M.; Leroy, C.; Nemazanyy, I.; Kuperwasser, N.; Pontoglio, M.; Friedlander, G.; Morel, E.; et al. The Primary Cilium and Lipophagy Translate Mechanical Forces to Direct Metabolic Adaptation of Kidney Epithelial Cells. Nat. Cell Biol. 2020, 22, 1091–1102.
- He, C.; Sumpter, R., Jr.; Levine, B. Exercise Induces Autophagy in Peripheral Tissues and in the Brain. Autophagy 2012, 8, 1548–1551.
- Claude-Taupin, A.; Codogno, P.; Dupont, N. Links between Autophagy and Tissue Mechanics. J. Cell Sci. 2021, 134, jcs258589.
- Mameli, E.; Martello, A.; Caporali, A. Autophagy at the Interface of Endothelial Cell Homeostasis and Vascular Disease. FEBS J. 2021, 289, 2976–2991.
- Madrigal-Matute, J.; de Bruijn, J.; van Kuijk, K.; Riascos-Bernal, D.F.; Diaz, A.; Tasset, I.; Martín-Segura, A.; Gijbels, M.J.J.; Sander, B.; Kaushik, S.; et al. Protective Role of Chaperone-Mediated Autophagy against Atherosclerosis. Proc. Natl. Acad. Sci. USA 2022, 119, e2121133119.
- Doronzo, G.; Astanina, E.; Corà, D.; Chiabotto, G.; Comunanza, V.; Noghero, A.; Neri, F.; Puliafito, A.; Primo, L.; Spampanato, C.; et al. TFEB Controls Vascular Development by Regulating the Proliferation of Endothelial Cells. EMBO J. 2019, 38, e98250.
- Shadab, M.; Millar, M.W.; Slavin, S.A.; Leonard, A.; Fazal, F.; Rahman, A. Autophagy Protein ATG7 Is a Critical Regulator of Endothelial Cell Inflammation and Permeability. Sci. Rep. 2020, 10, 13708.
- Reglero-Real, N.; Pérez-Gutiérrez, L.; Yoshimura, A.; Rolas, L.; Garrido-Mesa, J.; Barkaway, A.; Pickworth, C.; Saleeb, R.S.; Gonzalez-Nuñez, M.; Austin-Williams, S.N.; et al. Autophagy Modulates Endothelial Junctions to Restrain Neutrophil Diapedesis during Inflammation. Immunity 2021, 54, 1989–2004.e9.
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in Infection, Inflammation and Immunity. Nat. Rev. Immunol. 2013, 13, 722–737.
- Bharath, L.P.; Cho, J.M.; Park, S.-K.; Ruan, T.; Li, Y.; Mueller, R.; Bean, T.; Reese, V.; Richardson, R.S.; Cai, J.; et al. Endothelial Cell Autophagy Maintains Shear Stress–Induced Nitric Oxide Generation via Glycolysis-Dependent Purinergic Signaling to Endothelial Nitric Oxide Synthase. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1646–1656.
- Bharath, L.P.; Mueller, R.; Li, Y.; Ruan, T.; Kunz, D.; Goodrich, R.; Mills, T.; Deeter, L.; Sargsyan, A.; Anandh Babu, P.V.; et al. Impairment of Autophagy in Endothelial Cells Prevents Shear-Stress-Induced Increases in Nitric Oxide Bioavailability. Can. J. Physiol. Pharmacol. 2014, 92, 605–612.
- Zhang, J.-X.; Qu, X.-L.; Chu, P.; Xie, D.-J.; Zhu, L.-L.; Chao, Y.-L.; Li, L.; Zhang, J.-J.; Chen, S.-L. Low Shear Stress Induces Vascular ENOS Uncoupling via Autophagy-Mediated ENOS Phosphorylation. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Res. 2018, 1865, 709–720.
- Wang, S.; Iring, A.; Strilic, B.; Albarrán Juárez, J.; Kaur, H.; Troidl, K.; Tonack, S.; Burbiel, J.C.; Müller, C.E.; Fleming, I.; et al. P2Y2 and Gq/G11 Control Blood Pressure by Mediating Endothelial Mechanotransduction. J. Clin. Investig. 2015, 125, 3077–3086.
- Dela Paz, N.G.; Melchior, B.; Frangos, J.A. Shear Stress Induces Gαq/11 Activation Independently of G Protein-Coupled Receptor Activation in Endothelial Cells. Am. J. Physiol. -Cell Physiol. 2017, 312, C428–C437.
- Albarrán-Juárez, J.; Iring, A.; Wang, S.; Joseph, S.; Grimm, M.; Strilic, B.; Wettschureck, N.; Althoff, T.F.; Offermanns, S. Piezo1 and Gq/G11 Promote Endothelial Inflammation Depending on Flow Pattern and Integrin Activation. J. Exp. Med. 2018, 215, 2655–2672.
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The Role of Autophagy in the Heart. Annu. Rev. Physiol. 2018, 80, 1–26.
- Nakamura, M.; Sadoshima, J. Mechanisms of Physiological and Pathological Cardiac Hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407.
- Kanamori, H.; Takemura, G.; Goto, K.; Maruyama, R.; Tsujimoto, A.; Ogino, A.; Takeyama, T.; Kawaguchi, T.; Watanabe, T.; Fujiwara, T.; et al. The Role of Autophagy Emerging in Postinfarction Cardiac Remodelling. Cardiovasc. Res. 2011, 91, 330–339.
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct Roles of Autophagy in the Heart During Ischemia and Reperfusion. Circ. Res. 2007, 100, 914–922.
- Yan, L.; Vatner, D.E.; Kim, S.-J.; Ge, H.; Masurekar, M.; Massover, W.H.; Yang, G.; Matsui, Y.; Sadoshima, J.; Vatner, S.F. Autophagy in Chronically Ischemic Myocardium. Proc. Natl. Acad. Sci. USA 2005, 102, 13807–13812.
- García-Hoz, C.; Sánchez-Fernández, G.; García-Escudero, R.; Fernández-Velasco, M.; Palacios-García, J.; Ruiz-Meana, M.; Díaz-Meco, M.T.; Leitges, M.; Moscat, J.; García-Dorado, D.; et al. Protein Kinase C (PKC) ζ-Mediated Gαq Stimulation of ERK5 Protein Pathway in Cardiomyocytes and Cardiac Fibroblasts. J. Biol. Chem. 2012, 287, 7792–7802.
- Bartolomé, A.; García-Aguilar, A.; Asahara, S.-I.; Kido, Y.; Guillén, C.; Pajvani, U.B.; Benito, M. MTORC1 Regulates Both General Autophagy and Mitophagy Induction after Oxidative Phosphorylation Uncoupling. Mol. Cell. Biol. 2017, 37, e00441-17.
- Adams, J.W.; Pagel, A.L.; Means, C.K.; Oksenberg, D.; Armstrong, R.C.; Brown, J.H. Cardiomyocyte Apoptosis Induced by Gαq Signaling Is Mediated by Permeability Transition Pore Formation and Activation of the Mitochondrial Death Pathway. Circ. Res. 2000, 87, 1180–1187.
- Meleka, M.M.; Edwards, A.J.; Xia, J.; Dahlen, S.A.; Mohanty, I.; Medcalf, M.; Aggarwal, S.; Moeller, K.D.; Mortensen, O.V.; Osei-Owusu, P. Anti-Hypertensive Mechanisms of Cyclic Depsipeptide Inhibitor Ligands for Gq/11 Class G Proteins. Pharmacol. Res. 2019, 141, 264–275.
- Yun, C.W.; Jeon, J.; Go, G.; Lee, J.H.; Lee, S.H. The Dual Role of Autophagy in Cancer Development and a Therapeutic Strategy for Cancer by Targeting Autophagy. Int. J. Mol. Sci. 2020, 22, 179.
- Chude, C.; Amaravadi, R. Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. Int. J. Mol. Sci. 2017, 18, 1279.
- Filomeni, G.; de Zio, D.; Cecconi, F. Oxidative Stress and Autophagy: The Clash between Damage and Metabolic Needs. Cell Death Differ. 2015, 22, 377–388.
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of Tumorigenesis by Heterozygous Disruption of the Beclin 1 Autophagy Gene. J. Clin. Investig. 2003, 112, 1809–1820.
- Shen, Y.; Li, D.-D.; Wang, L.-L.; Deng, R.; Zhu, X.-F. Decreased Expression of Autophagy-Related Proteins in Malignant Epithelial Ovarian Cancer. Autophagy 2008, 4, 1067–1068.
- Peng, Y.-F.; Shi, Y.-H.; Ding, Z.-B.; Ke, A.-W.; Gu, C.-Y.; Hui, B.; Zhou, J.; Qiu, S.-J.; Dai, Z.; Fan, J. Autophagy Inhibition Suppresses Pulmonary Metastasis of HCC in Mice via Impairing Anoikis Resistance and Colonization of HCC Cells. Autophagy 2013, 9, 2056–2068.
- Chmurska, A.; Matczak, K.; Marczak, A. Two Faces of Autophagy in the Struggle against Cancer. Int. J. Mol. Sci. 2021, 22, 2981.
- Tsai, M.-J.; Chang, W.-A.; Huang, M.-S.; Kuo, P.-L. Tumor Microenvironment: A New Treatment Target for Cancer. ISRN Biochem. 2014, 2014, 351959.
- Das, J.; Maji, S.; Agarwal, T.; Chakraborty, S.; Maiti, T.K. Hemodynamic Shear Stress Induces Protective Autophagy in HeLa Cells through Lipid Raft-Mediated Mechanotransduction. Clin. Exp. Metastasis 2018, 35, 135–148.
- Lien, S.-C.; Chang, S.-F.; Lee, P.-L.; Wei, S.-Y.; Chang, M.D.-T.; Chang, J.-Y.; Chiu, J.-J. Mechanical Regulation of Cancer Cell Apoptosis and Autophagy: Roles of Bone Morphogenetic Protein Receptor, Smad1/5, and P38 MAPK. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Res. 2013, 1833, 3124–3133.
- Wang, K.; Wei, Y.; Liu, W.; Liu, L.; Guo, Z.; Fan, C.; Wang, L.; Hu, J.; Li, B. Mechanical Stress-Dependent Autophagy Component Release via Extracellular Nanovesicles in Tumor Cells. ACS Nano 2019, 13, 4589–4602.
- Yan, Z.; Su, G.; Gao, W.; He, J.; Shen, Y.; Zeng, Y.; Liu, X. Fluid Shear Stress Induces Cell Migration and Invasion via Activating Autophagy in HepG2 Cells. Cell Adhes. Migr. 2019, 13, 152–163.
- Follain, G.; Herrmann, D.; Harlepp, S.; Hyenne, V.; Osmani, N.; Warren, S.C.; Timpson, P.; Goetz, J.G. Fluids and Their Mechanics in Tumour Transit: Shaping Metastasis. Nat. Rev. Cancer 2020, 20, 107–124.
- Swartz, M.A.; Lund, A.W. Lymphatic and Interstitial Flow in the Tumour Microenvironment: Linking Mechanobiology with Immunity. Nat. Rev. Cancer 2012, 12, 210–219.
- Kalluri, R.; Zeisberg, M. Fibroblasts in Cancer. Nat. Rev. Cancer 2006, 6, 392–401.
- Goruppi, S.; Clocchiatti, A.; Dotto, G.P. A Role for Stromal Autophagy in Cancer-Associated Fibroblast Activation. Autophagy 2019, 15, 738–739.
- Kang, J.I.; Kim, D.H.; Sung, K.W.; Shim, S.M.; Cha-Molstad, H.; Soung, N.K.; Lee, K.H.; Hwang, J.; Lee, H.G.; Kwon, Y.T.; et al. P62-Induced Cancer-Associated Fibroblast Activation via the Nrf2-ATF6 Pathway Promotes Lung Tumorigenesis. Cancers 2021, 13, 864.
- Linares, J.F.; Cid-Diaz, T.; Duran, A.; Osrodek, M.; Martinez-Ordoñez, A.; Reina-Campos, M.; Kuo, H.-H.; Elemento, O.; Martin, M.L.; Cordes, T.; et al. The Lactate-NAD+ Axis Activates Cancer-Associated Fibroblasts by Downregulating P62. Cell Rep. 2022, 39, 110792.