Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Andrew Clayton and Version 2 by Conner Chen.
About one-third of the total protein targets in the pharmaceutical research sector are kinase-based. While kinases have been attractive targets to combat many diseases, including cancer, selective kinase inhibition has been challenging, because of the high degree of structural homology in the active site where many kinase inhibitors bind. Spectroscopic approaches such as infrared, Raman, NMR and fluorescence have the potential to provide significant insights into drug-target and drug-non-target interactions because of sensitivity to molecular environment.
- cancer
- anti-cancer drugs
- tyrosine kinase
1. What Is Fluorescence?
In order for peopleus to appreciate intrinsically fluorescent anti-cancer drugs, the reader is we first introducede the reader to the basics of fluorescence itself [1][14]. Fluorescence, a form of luminescence that can occur either in gas, liquid or solid chemical systems, is the emission of electromagnetic radiation, usually visible light, by a material that has absorbed light or other electromagnetic radiation. In the case of fluorescence, as shown in Figure 1a, the emitted light has a longer wavelength, i.e., lower photon energy compared to the absorbed radiation. This difference in wavelength between the positions of those band maxima is called the Stokes shift.
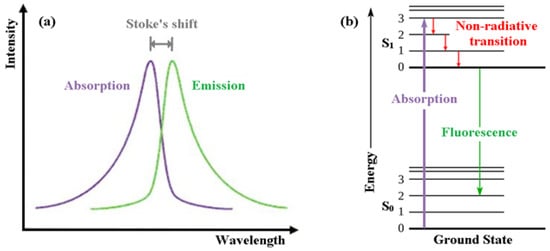
Figure 1.
(
a
) Emission intensity vs. wavelength plot. (
b
) Jablonski energy diagram for absorption and fluorescence.
The mechanism of fluorescence can be explained with the help of the Jablonski energy diagram (Figure 1b). The upward violet arrow represents the absorption of a photon in the singlet electronic ground state (So), causing a promotion of an electron to the singlet excited electronic state, S1. The downward red arrows denote vibrational relaxation from vibrationally excited states within the S1 manifold. This is a non-radiative relaxation process (i.e., no photon is emitted) in this case because the excitation energy is dispersed as vibrations or heat to the solvent. The downward green arrow denotes the fluorescence process from S1 to S0.
Note that because fluorescence occurs from the lowest vibrational level of the excited-state (S1) to higher vibrational levels of the ground-state (S0), the emission maximum of the fluorescence spectrum is always at lower energy (longer wavelength) than the excitation.
The quantum yield is an important parameter in fluorescence and its sensitivity to environment forms the basis for assays for detecting drug binding to drug targets. The quantum yield is defined as the number of photons emitted per photon absorbed and is in the range of 0 to 1. The quantum yield is determined by the relative rates of non-radiative and radiative processes that deplete the excited state. Radiative and non-radiative rates can be very dependent on environment, such as the binding sites of drug targets, so drug binding can be accompanied by large changes in quantum yield (or equivalently changes in amplitude of the fluorescence spectrum).
Another important fluorescence parameter is called the fluorescence lifetime. The fluorescence lifetime is the average time a molecule spends in the excited state. In contrast to quantum yield, which is related to relative rates of processes, the fluorescence lifetime is defined as the reciprocal of the sum of all the rates of processes depleting the excited state. Typical fluorescence lifetimes are in the range of 1–10 ns.
The finite lifetime of the excited state means that fluorescence can be sensitive to molecular structure and dynamics in the vicinity of the fluorophore. Myriad processes can occur during the excited state including rotation, collisions with other molecules, or a change in structure or conformation and environmental (solvent) relaxation.
Solutions containing the fluorophore are normally studied with a special spectrometer called a fluorometer, usually with a single exciting wavelength and variable detection wavelength (scanned to create a fluorescence spectrum). Because of the sensitivity that the method affords, fluorescent molecule concentrations as low as the nanomolar level can be measured [1][14]. Fluorescence in several wavelengths can be detected by an array detector, to detect compounds from high performance liquid chromatography (HPLC) flow. Thin layer chromatography (TLC) plates can also be visualized if the compounds or a coloring reagent is fluorescent. For visualizing fluorescence in cells, one uses a fluorescence microscope.
Molecular structure and chemical environment affect whether or not a substance undergoes fluorescence. When fluorescence does occur, molecular structure and local environment determine the color and intensity of emission. Hence, fluorescence can be used to investigate binding affinities, binding mechanisms, properties (i.e., polarity) of the binding site on the protein and binding kinetics. Another example is the effects of solvent on the structure and spectroscopic behavior of a fluorophore. Generally, solvatochromism is observed due to the differential solvation of the ground and excited states of a fluorophore. It was found that the optical spectroscopic measurements of a fluorophore can be influenced by the change in physicochemical properties of the surrounding medium. Solvatochromism is the term used to define this phenomenon and firstly introduced by Hantzschlater. The change in compound absorption/emission spectrum is manifested by one or more alternations in the band position, intensity or shape [2][3][4][15,16,17]. The hypsochromic (blue) shift of the fluorescence band relative to the absorption band is commonly known as negative solvatochromism. While positive solvatochromism is the term given for the bathochromic (red) shift of the fluorescence band [5][18], in negative solvatochromism, the molecule in its ground state is more stabilized than in the excited state upon increasing solvent polarity. When the excited state is more stabilized than the ground state, it results in a positive solvatochromism [5][18].
Generally, molecules that fluoresce are conjugated systems. Since most of the tyrosine kinase inhibitors are small aromatic molecules, their extended conjugation and ionizable groups render them good candidates for intrinsically fluorescent anti-cancer drugs.
2. Quinazoline-Based TKI
Quinazoline is one of the most widespread scaffolds among natural and synthetic bioactive compounds. Quinazoline scaffold resembles both the purine nucleus and the pteridine one. As a consequence, some compounds able to inhibit the purinic [6][19] or the folic acid [7][20] metabolic pathways have been discovered. Over the past few decades, the therapeutic potential of quinazoline derivatives has been found as different types of anticancer agents such as protein kinase inhibitors, tubulin polymerization inhibitors, protein lysine methyltransferase inhibitors, topoisomerase I inhibitors, PI3K/Akt/mTOR inhibitors, poly(ADP-ribose)polymerase-1 (PARP-1) inhibitors etc. [8][21]. Quinazoline ring is very commonly found in all types of tyrosine kinase inhibitors (TKIs) [9][22]. In general, quinazoline derivatives are known to possess a wide range of activities. A specific activity depends on the substituent present at an appropriate position of quinazoline.
For designing TKIs, 4-anilinoquinazolines found to be potent selective inhibitors [10][23]. The 4-anilinoquinazoline scaffold, i.e., the quinazoline core and N-aryl arm, together, form established ErbB/EGFR (epidermal growth factor receptor) pharharmacophore and used in type I (e.g., Gefitinib), type II (e.g., Lapatinib) and covalent inhibitors (e.g., Dacomitinib). This pharmacophore N-aryl arm is oriented deep for binding to the adenosine triphosphate (ATP)ATP nucleotide pocket [11][24]. The introduction of strong electron-donating or -withdrawing groups to the tail section being projected into the solvent produces donor–acceptor systems that are extremely sensitive to solvent polarity. The first- and second-generation EGFR–TKIs both have aniline-quinazoline structures. However, the second-generation TKIs also have an acrylamide group at 6 position of quinazoline ring, which serves as a chemically reactive electrophile called a Michael acceptor [12][26] that targets a cysteine nucleophile (Cys-797), resulting in a covalent adduct. Importance of fluorophore arm and pharmacophore arm of the 4-aminoquinazolines is well documented by J. Dhuguru et al. [11][24].
3. Basic Fluorescence Spectroscopy of TKIs
Solvatochromism is commonly used in many research aspects to characterize the nature of bulk environment [13][27]. Studying solvent effects on a fluorophore is commonly utilized to estimate the photophysical properties of a fluorophore. Fluorophore interaction with its environment can be triggered through specific interactions between fluorophore and solvent molecules, e.g., H-bond formation and non-specific interactions. Electrostatic interactions, driven by the change in solvent dipole moment, refractive index, relative permittivity (or dielectric constant), polarizability and viscosity, can induce a significant change in fluorophore electronic configuration and spectrum [14][28].
A number of empirical solvent models have been developed for the solvatochromic analysis of organic molecules. Linear solvation energy relationship (LSER) models have been proven to reliably define the interactions between solute and solvent molecules. Models developed by Bilot–Kawski [15][29] Lippert-Abboud–Mataga (L–M) [16][17][30,31], Bakhshiev [18][32] and Kawski–Chamma–Viallet [19][33] are among the most commonly used LSER models for solvatochromism analysis.
A summary of selected spectroscopic properties of TKI’s is collected in Table 1 [20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49].
Table 1.
Spectroscopic properties of selected kinase inhibitors.
| Drug (Intended Target) | Absorbance Max (nm) | Fluorescence Max (nm) |
|---|---|---|
| Sorafenib (Raf kinase) | (DCM/ACN) 1 264 nm | 487 nm [28][29][42,43] |
| Gefitinib (EGFR) | n-hexane (332 nm, 344 nm) | 368 nm, RI 7 = 1 [21][22][35,36] |
| Gefitinib (EGFR) | Benzene (332 nm, 340 nm) | 446 nm RI = 0.3 [21][22][35,36] |
| Gefitinib (EGFR) | n-octanol (334 nm, 344 nm) | 390–450 nm RI = 0.07 [21][22][35,36] |
| Gefitinib (EGFR) | Water (330 nm) | Non-fluorescent |
| Gefitinib (EGFR) | HSA (not reported) | 378 nm [21][22][35,36] |
| Erlotinib (EGFR) | n-hexane (332 nm, 344 nm) | 372 nm RI = 1 [22][23][36,37] |
| Erlotinib (EGFR) | Benzene (336 nm, 346 nm) | 423 nm RI = 0.38 [22][23][36,37] |
| Erlotinib (EGFR) | n-octanol (336 nm, 346 nm) | 400–470 nm RI = 0.07 [22][23][36,37] |
| Erlotinib (EGFR) | Water (332 nm) | Non-fluorescent [22][23][36,37] |
| Erlotinib (EGFR) | HAS 2 (336 nm, 347 nm) | 380–400 nm [22][23][36,37] |
| Lapatinib (EGFR, HER2) | THF 3 (380 nm) | 475 nm [24][38] |
| Lapatinib (EGFR, HER2) | Methanol (367 nm) | Non-fluorescent |
| Lapatinib (EGFR, HER2) | BSA 4 (361 nm) | 423 nm QY 8 = 0.07 [24][25][38,39] |
| Lapatinib (EGFR, HER2) | ErBB2 (368 nm) | 445 nm QY = 0.30 [24][25][38,39] |
| Lapatinib (EGFR, HER2) | Aggregates (371 nm) | 464 nm QY = 0.04 [24][25][38,39] |
| Afatinib | Methanol (246 nm, 340nm) | Not reported [32][46[33],47] |
| Imatinib (Abl kinase) | Water (281 nm) | 307nm [26][27][40,41] |
| Dacomitinib (mutated EGFR) | DMSO 5 (343 nm) | 500 nm 9 [30][31][44,45] |
| Nintedanib | PBS 6/DMSO (390 nm) | 482 nm [34][35][48,49] |
| Bosutinib (Abl kinase) | 350 nm | 480 nm [20][34] |
1 DCM/CAN is dichloromethane/acetontrile; 2 HSA is human serum albumin; 3 THF is tetrahydrofuran; 4 BSA is bovine serum albumin; 5 DMSO is dimethylsulphoxide; 6 PBS is phosphate-buffered saline; 7 RI indicates relative intensity; 8 QY = fluorescence quantum yield; 9 personal communication MD Lutful Kabir, Swinburne University.
The spectroscopy properties depend on the type of drug and also the solvent in which the spectrum is measured. In general, for all of the drugs listed, the absorption transitions all occur in the ultraviolet region of the spectrum. The emission spectra are somewhat more sensitive to drug type and particularly sensitive to environment. Specific examples are illustrated below.
Khattab et al. [36][50] have carried out a detailed examination of the absorption and fluorescence spectra of AG1478 in twenty-one solvents of different polarity and hydrogen-bonding strength. Solvatochromic analyses using different solvent models was undertaken to investigate the potential of AG1478 as a fluorescent reporter of its own environment. Fluorescence spectral analyses showed that hydrogen bonding from the solvent to AG1478 plays important role in solvatochromism, showing synergistic effect with solvent polarity in stabilizing the excited state. Fluorescence quantum yields were found to be influenced by solvent H-bond donor ability, being higher in aprotic than in protic solvents. Theirs work underscores the importance of polarity and hydrogen bonding environment on the spectroscopic properties of AG-1478 as well as helps in understanding the interaction of AG-1478 in vitro and in vivo. Moreover, AG-1478 shares the same molecular scaffold with the commercial anti-cancer drugs Erlotinib and Gefitinib [37][38][51,52].
Water molecules are frequently found in protein binding sites and can be important determinants for drug–protein intermolecular interactions and stability. Therefore, binding interactions between explicit water molecules and TKI may influence the nature and diversity of drug chemical structures and properties. To better understand the importance of hydration bonding and the specific solvation by water molecules, Khattab et al. [39][53] used fluorescence spectroscopy in acetonitrile–water solutions. A significant quenching of the AG1478 fluorescence with added water was observed and attributed to the formation of specific complexes between AG1478 and water molecules. Using a combination of computational chemistry with the spectroscopic measurements, the authors identified three potential sites for H-bond formation between AG1478 and water molecules. The computational models predicted that the extent of hydrogen bonding to the drug affected the distribution of twisted and planar drug conformations. A possible but yet to be further proven explanation of the water-induced fluorescence quenching is that the water induces a twisted form of the drug, which is non-fluorescent. TAs discussed next in this review, the environmental sensitivity of some of these drugs makes them useful reporters when applied to drug–protein complexes. For example, fluorescence when applied to drug–protein complexes can be used (in combination with computational chemistry) to infer drug conformation (from the fluorescence excitation spectrum) and deduce the polarity of the binding site (the fluorescence emission spectrum).