Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by PEDRO NUNEZ ABADES and Version 2 by Camila Xu.
Neurodegenerative diseases are characterized by gradually progressive, selective loss of anatomically or physiologically related neuronal systems that produce brain damage from which there is no recovery. Neurodegenerative diseases, bridging their clinical differences, are characterized by the gradual and irreversible damage and loss of specific neuronal networks.
- oxidative stress
- neurodegenerative diseases
- hyperexcitability
1. Introduction
Degenerative diseases of the central nervous system (CNS) impose substantial medical and public health burdens on populations throughout the world. The overwhelming rise in the prevalence of these disorders parallels the rapid increase in human lifespan. Neurodegenerative diseases, bridging their clinical differences, are characterized by the gradual and irreversible damage and loss of specific neuronal networks. Different brain areas affected will trigger different diseases, with the most common being Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS) and multiple sclerosis (MS) [1]. Among these neurodegenerative diseases AD is the most prevalent, causing between 60% and 80% of dementia cases globally. This disease, derived from an initial deterioration of the hippocampus and a posterior deterioration of the cortex and brainstem regions, is manifested by psychological, motor and specially memory alterations [2]. In the case of PD, the degeneration affects the dopaminergic neurons from the substantia nigra pars compacta, notably producing motor disorders such as bradykinesia, akinesia, stiffness, resting tremor and postural instability as well as non-motor manifestations such as depression, anxiety, sleep disorders, and cognitive and autonomic function alterations [3]. With a lower incidence (2–11 cases per 100,000 people) [4], ALS is the most common disease affecting the motoneurons, and it is triggered by the gradual, irreversible loss of the motoneurons located in the corticospinal tract. Although in its early stages is commonly manifested as muscular weakness, in 25% of patients ALS may be first manifested as bulbar symptoms, characterized by speech and swallowing difficulties. These symptoms constantly evolve until the event of the death of the patient, usually produced by respiratory arrest [5]. Finally, MS is the most prevalent non-traumatic disabling disease among the young adult population. It is an autoimmune inflammatory disease that has been classically attributed to a demyelinating pathology of the CNS. Although optic neuritis is the first symptom of MS for many people, the late stages of this disorder are accompanied with brain and spinal atrophy from which the brainstem and spinal cord syndromes derive [6]. All these neurodegenerative disorders seem to have common etiopathogenic pathways, albeit each of them affect different regions of the CNS, leading to different clinical manifestations. Thus, protein misfolding and aggregation as well as oxidative stress are highly recognizable common features in all these pathologies. In terms of neuronal functionality, hyperexcitability appears to be the common hallmark among all of them and often the degree of excitability of patient’s derived motor neurons correlates with the patient’s rate of survival, especially in ALS [7][8][9][7,8,9]. However, the molecular and cellular mechanisms that lead to the onset of these malfunctions and thus to the development of the disease are complex and still poorly understood. Therefore, an in-depth understanding of the mechanisms underlying the neurodegenerative processes is a necessary step in the development of new treatments capable of preventing the emergence of these diseases or, at least, of delaying their progression [1][10][11][1,10,11].
2. Pathogenesis of Neurodegenerative Disease: Oxidative Stress and Inflammation
Irreversible neuronal deterioration is the most common consequence of neurodegenerative diseases -either AD, PD, ALS, MS or others. Although the pathogenesis of these diseases is not fully understood, several neurological components have been already identified as responsible for the neuronal pathology such as increased protein aggregates, glial activation/inflammation, mitochondrial dysfunction, disbalance of neurotransmitters, autophagia or oxidative stress (Figure 1). A comprehensive review of the molecular features and histopathology of all of these pathogenic mechanisms in neurodegenerative diseases is beyond the scope of this resviearchw except for oxidative stress but it is worth highlighting several aspects of the role of neuroinflammation because of its interrelation with oxidative stress and hyperexcitability in motor cortex neurons.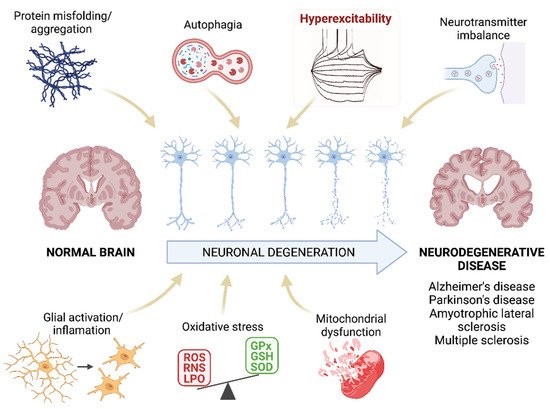
Figure 1. Pathogenesis of neurodegenerative diseases. Schematic representation of the mechanisms that are proposed as the main pathogenic agents of neurodegenerative diseases. ResWearchers highlight the hyperexcitability as this parameter will be the main focus of this researchview. ROS: reactive oxygen species; RNS: reactive nitrogen species; LPO: lipid peroxidation; GPx: glutathione peroxidase; GSH: glutathione; SOD: superoxide dismutase. Created with BioRender.com [12].
2.1. Inflammation Triggers Neurodegenerative Diseases
The blood-brain barrier (BBB) is the main protective barrier of the CNS. It has a highly selective permeability and, in addition of selectively allowing the entrance of essential nutrients such as glucose and amino acids, it also regulates the controlled access of immune cells into the brain. An increased BBB permeability to immune cells can disbalance the homeostasis and provoke serious neurological deficits. Thus, a common feature of these CNS diseases is a chronic immune activation, affecting innate and adaptative immune responses. In addition, microglia, astrocytes, endothelial cells, and peripheral immune cells release inflammatory cytokines and chemokines, causing a prolonged proinflammatory state of microglia, increasing the permeability of the BBB and facilitating the infiltration of immune cells into the CNS [13][14][13,14]. Microgliosis. Microglia composes the immune system of the CNS. In addition to their phagocytic capacity, active microglia releases proinflammatory molecules such as nitric oxide (NO), tumor necrosis factor α (TNFα), major histocompatibility complex class II (MHC II), prostaglandin-endoperoxide synthase 2 (Cox-2), monocyte chemotaxic protein 1 (MCP-1), and interleukins 1β and 6 (IL-1β and IL-6), playing a crucial role in the inflammatory response. Although microglia is necessary for the elimination of dead neurons, its overactivation is related to a faster progression of neurodegenerative diseases such as AD, PD, ALS and MS [15][16][17][18][19][20][15,16,17,18,19,20]. Under normal conditions, microglia is in a “resting” state morphologically characterized by ramifications that extend and retract so that these cells can constantly monitor the state of the microenvironment that surrounds them. When an abnormality is detected, the resting microglia transitions to an activated state in which the morphology changes to an ameboid shape. Active microglia can be observed, for example, surrounding senile plaques in AD. In addition to activation markers, this microglia expresses proinflammatory mediators such as MCP-1 that contribute to the recruitment of astrocytes in senile plaques [20]. In PD it has been suggested that microgliosis is caused by α-Syn aggregates, and that this active microglia contributes to the death of dopaminergic neurons [21]. Likewise, microgliosis is also a hallmark of neuroinflammation in ALS. Active microglia has been observed by positron emission tomography in the brains of living patients in which, the intensity of activation correlates with the severity of the disease, depicting the role of active microglia in the course of the disease [22]. Microglial activation is also evident in MS, both in early and later stages of the disease. Its activation correlates with axon and oligodendrocyte pathology, in which myelin remnants and cell fragments can be found. In addition to the production of various inflammatory neurotoxic mediators, microglia secretes the proteolytic lipolytic enzymes which ultimately lead to the destruction of myelin [23]. Reactive Astrocytes. Astrocytes are the main components of the glia and perform numerous functions of support and maintenance of the nervous system [24]. Activated astrocytes can give rise to two different forms, known as A1 and A2. While A2 favors tissue repair, A1 favors its degradation. The activation of the latter contributes to neuroinflammation through the release of proteins and inflammatory mediators. Thus, in neurodegenerative diseases astroglia lose its protective function which may cause the collapse of the BBB and neuronal death. For example, in the case of AD, astroglia A1 causes an imbalance in the release of calcium, glutamate and GABA, contributing to the development of the disease [25]. In fact, in many cases the cognitive impairment associated with AD correlates with the number of reactive astrocytes present in senile plaques and with the concentration of proinflammatory mediators produced by them [26]. Moreover, A1 astrocytes promote the formation of amyloid beta (Aβ) plaques [24]. A1 reactivity has also been detected in the substantia nigra of PD patients [27]. At the beginning of the disease, α-Syn inclusions are found inside the astrocytes (also increased in number), which leads to the mobilization of phagocytic microglia and the destruction of cholinergic neurons, producing the clinical symptoms of the disease [28]. Similarly, the degree of astrocytic reactivity correlates directly with the rate of neuronal death in ALS [24][29][24,29]. Finally, the existence of reactive astrocytes has also been demonstrated in MS, being the main source of proinflammatory cytokines. These cytokines are the cause of uncontrolled inflammatory reactions with T cell infiltration and associated damage [24][30][31][24,30,31]. Pattern-recognition receptors (PRRs). Innate immune responses are triggered by PRRs. The best known PRRs are the Toll-like receptors (TLRs). It has been proved that CNS cells can activate the innate immune system via motifs related to damage and/or stress, to which TLRs bind. In fact, several studies show that TLRs are upregulated in the brains of patients with neurodegenerative disorders such as AD, PD, ALS or MS [32][33][34][32,33,34]. For example, TLR2, TLR3, and TLR4 are overexpressed in microglia of patients with PD and ALS, activating pathways that lead to the production of proinflammatory cytokines that contribute to neuronal damage [13]. Likewise, the expression of TLR2 and TLR4 is also increased in AD, and it has been shown in vitro that Aβ induces TLR synthesis. However, this increase could also play a beneficial role in neurodegenerative diseases, since TLRs help the absorption and elimination of protein aggregates, including Aβ [35][36][37][35,36,37]. Thus, these receptors seem to play a dual role in the development of neurodegenerative disorders, being able to cause neuronal damage but also favoring tissue regeneration. This suggests that enhancing their beneficial functions may be useful as a therapeutic strategy in the treatment of these pathologies [13].2.2. Role of Oxidative Stress in Neurodegenerative Diseases
Another essential mechanism in neurodegenerative processes is oxidative stress [38]. Oxidative stress is known as the cellular situation in which the free radicals -reactive oxygen and/or nitrogen species (ROS and RNS, respectively)- exceed the natural antioxidant defenses of the organism [39]. The importance of these mechanisms in the development of AD, PD, ALS, and MS lies in the fact that neurons are especially vulnerable to ROS and RNS since they contain high levels of unsaturated lipids susceptible to oxidation and high levels of transition metals that catalyze the synthesis of free radicals [40]. Besides, the CNS has a high metabolic rate and, hence, a high oxygen demand which favors free radical formation. The metabolism of neurotransmitters is also a source of free radicals in the CNS. In addition, the antioxidant defense of the CNS is weaker than in other organs making it even more susceptible to oxidative stress. Under oxidative stress conditions, dysfunctional mitochondria are unable to produce the high energy levels required by neuronal cells to perform their normal biochemical and physiological functions. This mitochondrial impairment makes them vulnerable to rapid cell death [41]. Under physiological concentrations ROS play a crucial role as regulators of various cellular functions such as growth, proliferation, differentiation and survival; they participate in cognitive function and in the maintenance of antioxidant mechanisms [42][43][42,43]. However, an excess of ROS and/or RNS can damage cells modifying lipids, proteins, and DNA [38]. ROS produced by mitochondria cause lipid peroxidation (LPO), which alters the cell membrane structure, and integrity affecting cell signaling. In here, rwesearchers review the evidences that demonstrate the presence of oxidative stress in neurodegenerative diseases and its impact in AD, PD, MS and ALS (Figure 2).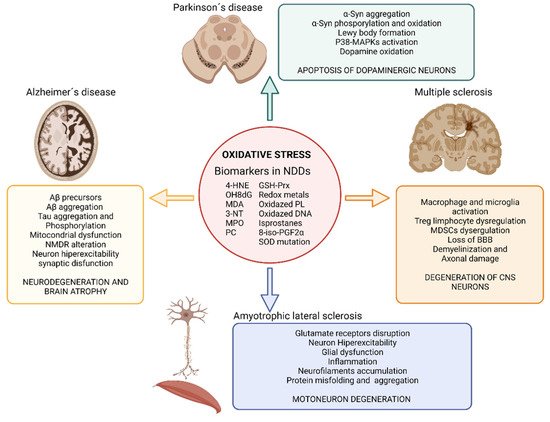
Figure 2. Impact of oxidative stress on neurodegenerative diseases. Illustration showing the impact of oxidative stress on Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis and multiple sclerosis and the main biomarkers of oxidative stress found in neurodegenerative disease’s studies. Abbreviatures: BBB, blood brain barrier; GSH-Prx, glutathione-peroxiredoxins; 4-HNE, 4-hidroxinonenal; NDDs, neurodegenerative diseases; NMDR, N-metil-D-aspartato receptors; MDA, malondialdehyde; MAPKs, Mitogen-activated protein kinases; MPO, Myeloperoxidase; 3-NT, 3-nitrotyrosine; OH8dG, 8-hydroxy-2′-deoxyguanosine; PC, protein carbonyl; 8-iso-PGF2α,8-iso-prostaglandin F2α; α-Syn, α-Synuclein; SOD, superoxide dismutase. Created with BioRender.com [12].