Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Lindsay Dong and Version 1 by François Michel Carlier.
Chronic obstructive pulmonary disease (COPD), asthma and cystic fibrosis (CF) are distinct respiratory diseases that share features such as the obstruction of small airways and disease flare-ups that are called exacerbations and are often caused by infections. Along the airway epithelium, immunoglobulin (Ig) A contributes to first line mucosal protection against inhaled particles and pathogens. Dimeric IgA produced by mucosal plasma cells is transported towards the apical pole of airway epithelial cells by the polymeric Ig receptor (pIgR), where it is released as secretory IgA. Secretory IgA mediates immune exclusion and promotes the clearance of pathogens from the airway surface by inhibiting their adherence to the epithelium.
- immunoglobulin A
- mucosal immunity
- COPD
1. Introduction
Each breath carries thousands of particles towards the airways, constituting many potential threats to lung integrity. Airway mucosal immunity provides both innate and adaptive responses against these inhaled agents, providing inflammatory responses against harmful antigens and tolerogenic mechanisms towards innocuous ones. Failure to these duties may lead to increased antigen penetration and recurrent infections or exaggerated immune responses towards harmless antigens, both situations potentially resulting in chronic airway inflammation. Immunoglobulin (Ig) A represents the predominant Ig at mucosal surfaces in humans [1], where it predominantly lays in its secretory form (S-IgA). Carried towards the airway lumen by its epithelial transporter, the polymeric Ig receptor (pIgR), S-IgA plays a crucial role in the immune exclusion of inhaled pathogens while IgA also regulates immune cells residing in or attracted to mucosal tissues [2].
2. The Mucosal S-IgA System in Homeostasis
The mucosal barrier system is constitutively made of several components. The epithelial layer and the intercellular junctions, with the mucus layer on top, and the lamina propria underneath [13[3][4],14], form a physical barrier against exogenous antigens and pathogens. Mucosal antimicrobial molecules, cellular innate immunity and mucosal adaptative immunity are complementary lines of defence against aggressions [15][5]. In the bronchi, the epithelium is pseudostratified, and composed by numerous cell types whose proportions are tightly controlled [16,17][6][7]. Most of the bronchial airway epithelium consists of ciliated and goblet cells that together constitute the mucociliary elevator clearing particulates and other irritants out of the airspaces. As well as these cell types, the airway epithelium also comprises basal, club and neuroendocrine cells, along with rare ionocytes [18][8]. Ciliated cells are prominent and represent more than 50% of all airway epithelial cells. They possess around 300 cilia whose synchronized beating pushes the mucus layer towards the trachea and the larynx [19][9]. Goblet cells, accounting for 5 to 15% of airway epithelial cells in health, produce mucus and are virtually not present in small airways [19][9]. Basal cells are multipotent stem cells that both anchor the epithelium to the underlying lamina propria and drive epithelial homeostasis and orderly regeneration after injury [20,21][10][11]. They represent 5 to 30% of epithelial cells, their proportion decreasing from the trachea down to the respiratory bronchioles [22][12]. Club cells are dome-shaped cells involved in host defence, and represent 20% of epithelial cells in small airways, where they also behave as progenitor cells [23,24][13][14]. Neuroendocrine cells are rare, innervated cells (< 1% of airway epithelial cells) [25][15] that are thought to be involved in oxygen sensing, smooth muscle tonus and immune responses [26,27][16][17]. Finally, recently discovered ionocytes seem to control the airway surface liquid and mucus viscosity [28][18]. Among these cell types, ciliated, goblet and club cells have been robustly shown to express the polymeric immunoglobulin receptor (pIgR) and therefore participate in the epithelial transcytosis of dimeric IgA (d-IgA) towards the apical mucus layer (see below) [29[19][20],30], while recent single cell transcriptomic data and the emergence of the Human Lung Atlas suggest that ionocytes (but not basal and neuroendocrine cells) also express the pIgR [31][21].2.1. Production and Structure of S-IgA and pIgR
IgA constitutes the most prevalent Ig isotype at mucosal sites and the second most prevalent in serum after IgG. Therefore, IgA is the most abundantly produced Ig in the human body, with an average production rate of 66 mg/kg/day [32,33][22][23]. IgA largely mediates the adaptive humoral immune defence at mucosal surfaces [34,35][24][25], while its role in serum remains relatively unexplored. IgA is produced by B cells both in the systemic and mucosal immune systems, the latter being referred to as mucosa-associated lymphoid tissue (MALT) which includes mucus layers, epithelial cells, lymphoid tissues and immune molecules of the mucosal lamina propria [36][26]. Structurally, IgA is found as monomers (m-IgA) or polymers, mainly consisting of dimers (d-IgA), although some larger forms also exist. M-IgA is composed of two light chains κ and λ, common to all types of Ig’s, covalently linked to two specific heavy chains α [37][27]. In serum, IgA represents 6 to 15% of total immunoglobulins [38][28], and predominates as monomers (85% to 90%). Serum IgA mainly originates from the bone marrow, although the spleen and peripheral lymph nodes could contribute to a lower extent [38][28]. In humans, two IgA subclasses exist, namely IgA1 and IgA2. Each of these IgA subtypes can be found in a monomeric, dimeric, or secretory form [40][29]. In bronchial secretions, IgA2 accounts for 30% of total IgA, compared to 10% in plasma [40][29]. B cells that secrete IgA in the airways are antibody-secreting plasma cells that are generated following cognate interactions between T cells and dendritic cells that have taken up and processed a specific antigen [42,43][30][31].2.2. Transcytosis of d-IgA and Functions of S-IgA
pIgR-mediated polymeric Ig transcytosis requires four well-orchestrated steps [51][32]. First, d-IgA (or pentameric IgM) binds to pIgR through a non-covalent interaction between Ig’s J chain and the extracellular part of pIgR. Second, pIgR is internalized in the clathrin-mediated endocytosis process and delivered in basolateral early endosomes, then in common endosomes. Endocytosed pIgR is then either recycled back to the basolateral membrane, or delivered to the apical surface [50][33]. Third, the extracellular part of the receptor (SC, bound to the transported Ig), undergoes endoproteolytic cleavage by a host serine proteinase. Lastly, S-IgA consisting of d-IgA and SC, crosses the mucus layer by diffusion. Of note is that, unbound pIgR may also undergo epithelial transcytosis and proteolytic cleavage, thereby releasing free SC that may be found in mucosal secretions [50,51][32][33]. At mucosal surfaces, S-IgA functions include neutralization, as well as other biological effects through interactions with, and regulation of, immune cells (via specific Fcα receptors) and microbiota components (Figure 1). The first-line mucosal defence is primarily exerted by S-IgA through its binding of soluble or particulate antigens, preventing their adherence to the epithelium. This non-specific immunity process called ‘immune exclusion’ consists of several sub-steps, including agglutination and entrapment as well as clearance at mucosal surfaces [2,53][2][34].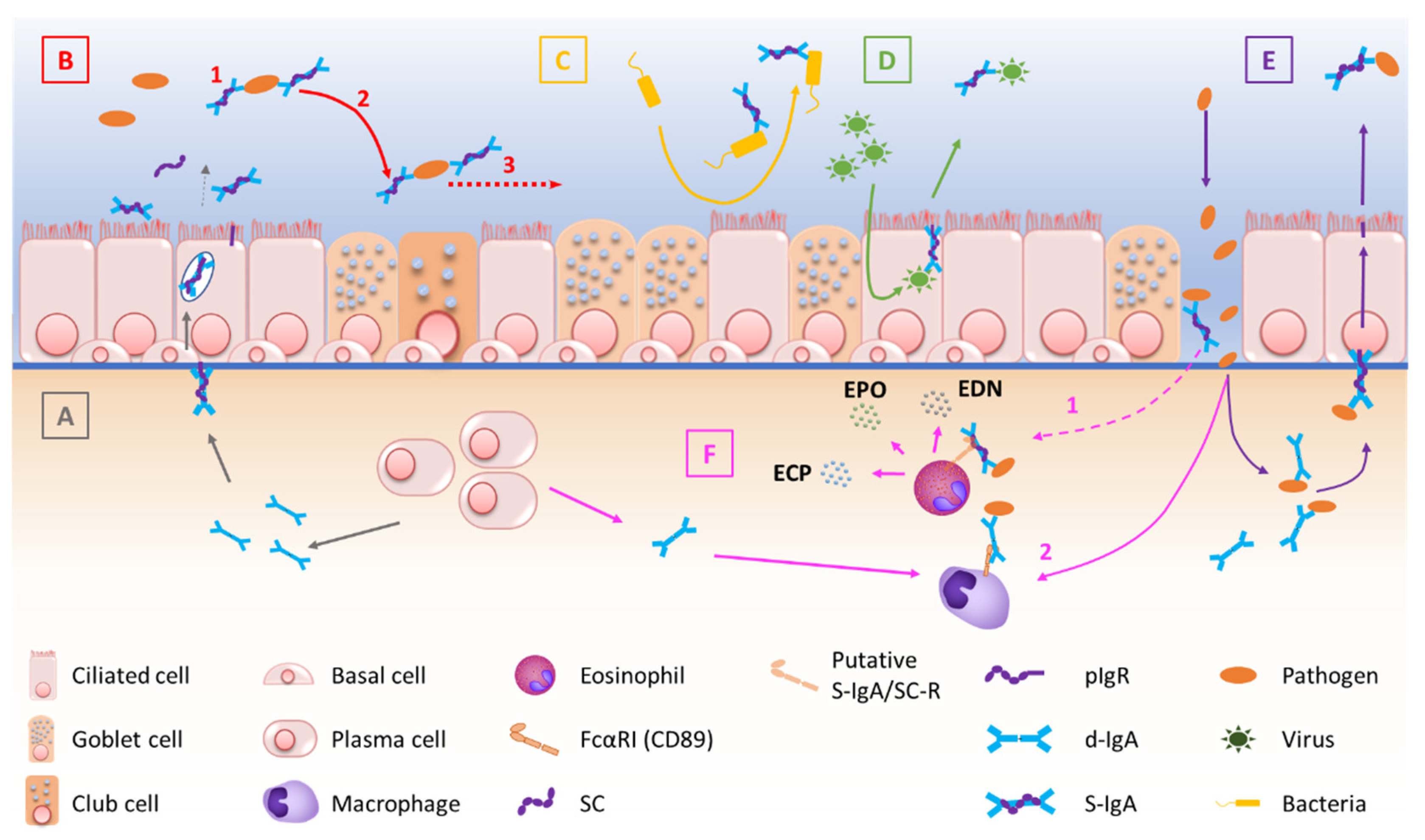
Figure 1. Schematic description of the multiple functions of IgA and the pIgR/S-IgA system at mucosal surfaces. (A) pIgR-mediated endosomal transcytosis of d-IgA, produced by submucosal B cells. (B) S-IgA-driven immune exclusion, including agglutination (1), entrapment of immune complexes (2) and clearance of trapped pathogens (3). (C) Inhibition of bacterial adherence to the mucosal epithelium. (D) Intraepithelial neutralization of penetrating viral antigens. (E) pIgR-mediated elimination of subepithelial immune complexes, after immune exclusion of pathogens by subepithelial d-IgA. (F) (1) S-IgA-induced degranulation of eosinophils resulting from binding of S-IgA to its eosinophil receptor (potentially FcαRI or another receptor such as C-type lectin), occurring possibly after epithelial barrier disruption and releasing eosinophil granule proteins. (2) IgA-induced engagement of phagocytes, enhancing clearance mechanisms.
2.3. Regulation of S-IgA Production
First, the PIGR gene promoter displays binding sites for inflammation-related factors such as IFN regulatory factor 1 (IRF-1), STAT6 and Nuclear Factor (NF)-κB [52][35]. Therefore, host cytokines that activate pathways involving STAT, IRF or NF-κB, such as TNF-α, IFN-ɣ, IL-4 and IL-1, are able to upregulate pIgR expression and d-IgA transepithelial routing [51,52,68][32][35][36]. Depending on the studies, however, exposure to inflammatory stimuli provides divergent results. For instance, IL-4 may stimulate pIgR expression in Calu-3 cell line cultures [69][37], while it inhibits pIgR expression in primary airway epithelial cells [70][38], contributing to pIgR downregulation found in the airway epithelium of asthma patients. A similar dual effect in cell line versus primary cells was observed with TGF-β1, as exogenous exposure of Calu-3 cells to TGF-β1 increases SC production, whereas pIgR production is inhibited by TGF-β1 in primary human bronchial epithelial cells [68,71][36][39]. The molecular substratum for such discrepancies remains unknown. In addition, inflammatory cytokines contribute to pIgR downregulation both in asthma and COPD, while IL-17 conversely upregulates pIgR in Pseudomonas aeruginosa (Pa) infected CF cells [68,70,72,73][36][38][40][41].
Finally, environmental factors may also influence pIgR expression. For instance, pIgR mRNA levels are increased in ex-smokers’ lungs. This increase is however not observed at the protein level, or in situ nor in air/liquid interface cultures of primary human bronchial epithelial cells exposed to cigarette smoke (CS) or derived from smokers [68,74][36][42]. These data suggest that the CS exposure acts as a player in the regulation of pIgR gene expression in vivo, which is further submitted to post-transcriptional modifications that have not been much explored so far. In addition, the microbiota also regulates pIgR through the release of microbe-associated molecular patterns (MAMPs), which control PIGR gene transcription through the activation of Toll-like receptors (TLR) [51,52][32][35].
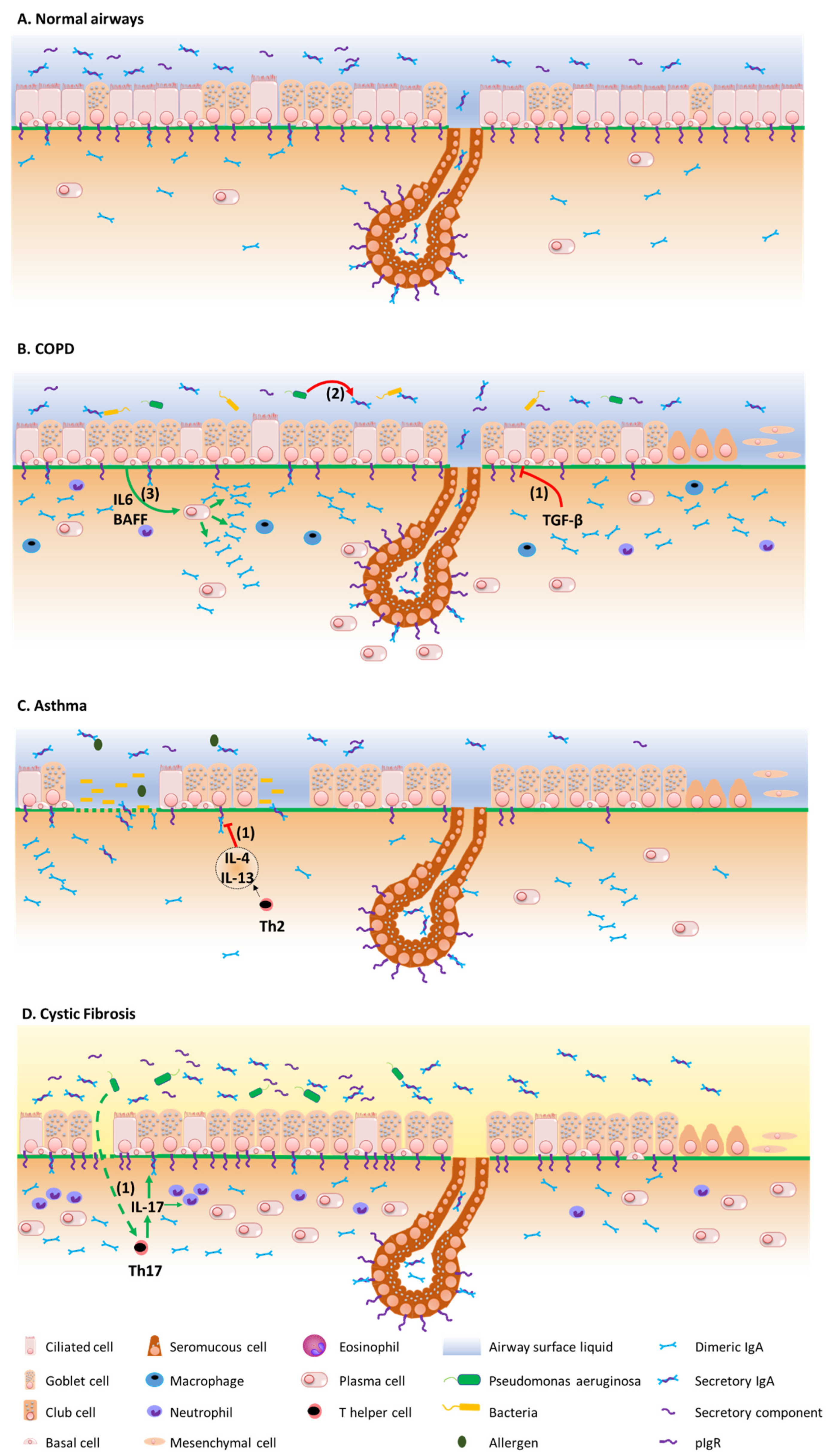
3. The Mucosal S-IgA System in Airway Disease
Figure 2 summarizes the mechanisms by which the IgA/pIgR system is altered in chronic respiratory diseases. Although resulting from complex physiopathological mechanisms in these diseases, respiratory tract colonization by pathogens and trespassing of the mucosal barrier have been shown to trigger these diseases, demonstrating their contribution to the development of such diseases [75,76][43][44].Figure 2. Overview of the pIgR/IgA system dysregulation mechanisms in chronic respiratory diseases. (A) pIgR/IgA system in airway homeostasis, at the epithelial surface and in submucosal glands. (B) In COPD, TGF-β induces pIgR downregulation at the epithelial surface (1), while pIgR expression is preserved in the submucosal glands. The S-IgA local deficiency relates to the subsequently decreased IgA transcytosis, as well as to S-IgA proteolysis by pathogen-derived proteinases (2), favouring bacterial invasion and innate immune cell infiltration. Subepithelial IgA may accumulate as a result of decreased transepithelial transport and IL-6- and BAFF-driven IgA synthesis (3). Enhanced survival of IgA+ plasma cells around the submucosal glands could contribute to a preserved S-IgA production at this level. (C) In asthma, IL-4/IL-13 may induce pIgR downregulation, also leading to luminal S-IgA deficiency (1). (D) In CF, pIgR is conversely upregulated, along with increased production of IgA and S-IgA in airway tissues and lumen, possibly through chronic infection by Pseudomonas aeruginosa that drives pIgR upregulation through IL-17 (1).
3.1. COPD
The functionality of the IgA/pIgR system in respiratory diseases was first explored in COPD, where the abundant literature now clearly demonstrates its multifaceted alteration [77][45]. Chronic obstructive pulmonary disease (COPD), currently representing the third leading cause of death worldwide [3][46], is mainly due to CS with potential additional contributions of other toxics (biomass, occupational, air pollution) and genetic predisposition [78][47].
In 2001, oura team first showed that pIgR/SC expression is decreased in the epithelium of large and small airways from COPD patients, as compared with both non-smokers and non-COPD smokers. In addition, SC expression inversely correlates with COPD severity and related functional parameters such as FEV1, FVC and MEF50 [81][48]. These observations were later corroborated in a larger cohort demonstrating that pIgR epithelial expression is mainly decreased in the airway epithelium from severe COPD patients, as compared with non-smoker controls, non-COPD smokers and less severe COPD patients, and associated with a persistence of the defect in primary cultures of bronchial epithelial cells from such patients where the mechanism could be shown as involving TGF-β signalling [68][36]. Recently, a genome-wide association study showed that PIGR gene expression was reduced in airway intermediate and ciliated cells from smokers without COPD [82][49]. Interestingly, pIgR downregulation in COPD is more obvious in zones of bronchial epithelial remodelling, such as goblet cell hyperplasia, squamous metaplasia, or incompletely differentiated areas. Accordingly, these specific zones also display S-IgA deficiency [83][50], contributing to localized reduced mucosal immunity, with those areas also exhibiting increased bacterial invasion, macrophage and neutrophil infiltration, as well as NF-κB activation [84][51].
3.2. Asthma
As with COPD, asthma is a broad clinical entity that manifests with symptoms of wheezing, shortness of breath, cough, and chest tightness. Asthma is characterized by reversible airflow obstruction, in contrast with COPD where the airflow obstruction is (mostly) irreversible [97][52]. Behind this clinical definition, asthma is a heterogeneous disease that relates to diverse pathophysiological mechanisms. Two main immune phenotypes are described, namely type 2 (T2-) high asthma, which includes several subsets according to the age of onset, allergic background, and comorbidities such as aspirin sensitivity and/or nasal polyposis; T2-low asthma, which includes obesity-related asthma [97,98,99,100][52][53][54][55]. Both early-onset allergic (or extrinsic) asthma and some forms of late-onset non-allergic asthma are characterized by a T2-mediated eosinophilic inflammation, whose underlying upstream mechanisms include epithelial activation by viruses, allergens and/or air pollution. The subepithelial penetration of these irritants is further enhanced by epithelial barrier dysfunction that is due to the decreased tightness of apical junctional complexes, as well as zones of epithelial shedding [77][45]. These airborne stimuli induce epithelial cells necrosis leading to the release of alarmins such as TSLP, IL-33, IL-25 and GM-CSF, the latter attracting and/or activating mast cells, basophils and eosinophils [51][32]. In T2-low asthma, macrophages recruitment is also driven by the epithelial release of CCL2 and CCL3 [101][56], while in obesity-related asthma, leptin activates airway epithelial cells and induces the production of cytokines such as IL-8, further contributing to neutrophil recruitment [101][56]. In turn, these inflammatory cells mediate airway remodelling, including goblet cells metaplasia, epithelial-to-mesenchymal transition and reticular basement membrane thickening, along with bronchial hyperresponsiveness and smooth muscle hypertrophy [51[32][53],98], as well as IgE class switch recombination in B cells [51][32]. In addition the increased levels of allergen specific IgE, higher IgA levels have been observed in the airway mucosa of allergic asthma patients [102,103,104][57][58][59]. Although the exact role(s) of S-IgA in the development of allergic disorders remains controversial, several lines of evidence suggest that the S-IgA system could play a protective role in asthma and allergic diseases [2]. Thus, higher levels of S-IgA in breast milk were associated to a lower risk of atopic dermatitis up to the age of two [108][60], and subjects with selective IgA deficiency display higher risks of developing allergic diseases [109][61]. In addition, populations with higher serum IgA levels display lower rates of house dust mite sensitization and severe airway hyperresponsiveness [110][62]. Conversely, a positive correlation exists between serum IgA specifically directed against cow’s milk β-lactoglobulin and wheat gliadin at the age of one, and IgE sensitization at the age of 6 [111][63].3.3. Cystic Fibrosis
In contrast with asthma and COPD [128][64], data concerning IgA in cystic fibrosis (CF) are scarcer. CF is a multisystemic disease mainly affecting the respiratory, digestive and reproductive tracts [129][65]. Chronic pulmonary infections, notably due to Pa, play a crucial role in the prognosis of the disease and constitute a major immune challenge for the airway epithelium [130][66]. Recently, we undertook one study assessing the pIgR/IgA system in a multimodal project, showing increased epithelial IgA+ B cells, IgA production and pIgR expression in the airway tissues, sputum, and serum from CF patients [72,73][40][41]. In addition, a mice model mimicking chronic lung infection by using Pa-coated microbeads instilled in the mice airways showed that infection could upregulate pIgR expression and IgA production in the lungs of F508del mice, partly in an IL-17-dependant manner [72][40]. This was observed in vivo although in vitro, CF-derived airway epithelial cells displayed reduced pIgR expression, that appeared to be related to the activation unfolded protein response. In parallel, pIgR transcytosis is enhanced in CF, as evidenced by increased SC concentration in CF sputum, although SC seems to be dysfunctional in CF, notably preventing its neutralization capacity of IL-8/CXCL-8 due to an altered glycosylation pattern [131][67]. Increased IgA concentration has been found in the serum from CF patients, as compared with controls [132][68], in particular in chronically Pa infected patients [133][69] or with high clinical severity according to Shwachman Kulczycki scores [134][70]. Similarly, increased IgA concentrations were found in BALF from CF patients as compared with controls [135][71].3.4. Bronchiolitis Obliterans Syndrome
Chronic lung allograft dysfunction (CLAD) represents a major hurdle in lung transplantation (LT), that burdens long-term survival [137][72]. Depending on pulmonary function tests and computed tomography, CLAD is currently separated into obstructive CLAD (first described as bronchiolitis obliterans syndrome (BOS), restrictive CLAD (also known as restrictive allograft syndrome (RAS), mixed CLAD (combining BOS and RAS features) and undefined CLAD [138][73]. BOS is the most frequent form of CLAD and is histologically characterized by epithelial injury, bronchocentric mononuclear inflammation, and fibrosis of small airways [144][74]. As repetitive and/or chronic infections have widely been associated with CLAD risk [145[75][76][77],146,147], it can be questioned whether impaired airway mucosal immunity (and thus reduced microbial eviction) could favor CLAD/BOS. However, the literature in the field is scarce, with no study so far assessing the airway expression of pIgR after LT. One previous study assessed BALF S-IgA levels in LT recipients, showing reduced S-IgA levels during so-called “rejection episodes” [148][78]. Of note is that, CLAD phenotypes had not been described at the time this study was published. More recently, Vandermeulen and colleagues showed increased total BALF IgA levels in LT recipients with RAS and BOS as compared with control LT recipients [149][79]. Finally, low serum IgA levels prior to LT represent a risk factor for developing post-LT infections [150[80][81],151], while lower post-LT IgA serum levels have been observed in patients with BOS as compared with control LT recipients [151][81].References
- Corthésy, B. Multi-Faceted Functions of Secretory IgA at Mucosal Surfaces. Front. Immunol. 2013, 4, 185.
- Carlier, F.M.; Sibille, Y.; Pilette, C. The epithelial barrier and immunoglobulin A system in allergy. Clin. Exp. Allergy 2016, 46, 1372–1388.
- Yang, S.; Yu, M. Role of Goblet Cells in Intestinal Barrier and Mucosal Immunity. J. Inflamm. Res. 2021, 14, 3171–3183.
- Boyaka, P.N.; Fujihashi, K. Host Defenses at Mucosal Surfaces. In Clinical Immunology, 5th ed.; Rich, R.R., Fleisher, T.A., Shearer, W.T., Schroeder, H.W., Frew, A.J., Weyand, C.M., Eds.; Elsevier: London, UK, 2019; pp. 285–298.e1.
- McNabb, P.C.; Tomasi, T.B. Host Defense Mechanisms at Mucosal Surfaces. Annu. Rev. Microbiol. 1981, 35, 477–496.
- Davis, J.D.; Wypych, T.P. Cellular and functional heterogeneity of the airway epithelium. Mucosal Immunol. 2021, 14, 978–990.
- Gohy, S.; Hupin, C.; Ladjemi, M.Z.; Hox, V.; Pilette, C. Key role of the epithelium in chronic upper airways diseases. Clin. Exp. Allergy 2019, 50, 135–146.
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020, 587, 619–625.
- Knight, D.A.; Holgate, S.T. The airway epithelium: Structural and functional properties in health and disease. Respirology 2003, 8, 432–446.
- Rock, J.R.; Randell, S.H.; Hogan, B.L.M. Airway basal stem cells: A perspective on their roles in epithelial homeostasis and remodeling. Dis. Model. Mech. 2010, 3, 545–556.
- Hogan, B.L.; Barkauskas, C.E.; Chapman, H.A.; Epstein, J.A.; Jain, R.; Hsia, C.C.; Niklason, L.; Calle, E.; Le, A.; Randell, S.H. Repair and regeneration of the respiratory system: Complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell 2014, 15, 123–138.
- Boers, J.E.; Ambergen, A.W.; Thunnissen, F.B.J.M. Number and Proliferation of Basal and Parabasal Cells in Normal Human Airway Epithelium. Am. J. Respir. Crit. Care Med. 1998, 157, 2000–2006.
- Zuo, W.-L.; Shenoy, S.A.; Li, S.; O’Beirne, S.L.; Strulovici-Barel, Y.; Leopold, P.L.; Wang, G.; Staudt, M.R.; Walters, M.S.; Mason, C.; et al. Ontogeny and Biology of Human Small Airway Epithelial Club Cells. Am. J. Respir. Crit. Care Med. 2018, 198, 1375–1388.
- Hiemstra, P.S.; Bourdin, A. Club cells, CC10 and self-control at the epithelial surface. Eur. Respir. J. 2014, 44, 831–832.
- Boers, J.E.; Brok, J.L.D.; Koudstaal, J.; Arends, J.W.; Thunnissen, F.B. Number and proliferation of neuroendocrine cells in normal human airway epithelium. Am. J. Respir. Crit. Care Med. 1996, 154, 758–763.
- Linnoila, R.I. Functional facets of the pulmonary neuroendocrine system. Lab. Investig. 2006, 86, 425–444.
- Branchfield, K.; Nantie, L.; Verheyden, J.M.; Sui, P.; Wienhold, M.D.; Sun, X. Pulmonary neuroendocrine cells function as airway sensors to control lung immune response. Science 2016, 351, 707–710.
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018, 560, 319–324.
- He, W.-H.; Zhang, W.-D.; Cheng, C.-C.; Lu, J.; Liu, L.; Chen, Z.-H.; Wang, W.-H. Expression characteristics of polymeric immunoglobulin receptor in Bactrian camel (Camelus bactrianus) lungs. PLoS ONE 2022, 17, e0264815.
- Blackburn, J.B.; Schaff, J.A.; Gutor, S.; Du, R.-H.; Nichols, D.; Sherrill, T.; Gutierrez, A.J.; Xin, M.K.; Wickersham, N.; Zhang, Y. Secretory cells are the primary source of pIgR in small airways. bioRxiv 2021.
- Schiller, H.B.; Montoro, D.T.; Simon, L.M.; Rawlins, E.L.; Meyer, K.B.; Strunz, M.; Braga, F.A.V.; Timens, W.; Koppelman, G.H.; Budinger, G.R.S.; et al. The Human Lung Cell Atlas: A High-Resolution Reference Map of the Human Lung in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2019, 61, 31–41.
- Woof, J.M.; Russell, M.W. Structure and function relationships in IgA. Mucosal Immunol. 2011, 4, 590–597.
- Bakema, J.E.; van Egmond, M. Immunoglobulin A: A next generation of therapeutic antibodies? mAbs 2011, 3, 352–361.
- Monteiro, R.C. The Role of IgA and IgA Fc Receptors as Anti-Inflammatory Agents. J. Clin. Immunol. 2010, 30, 61–64.
- Underdown, B.J.; Schiff, J.M. Immunoglobulin A: Strategic Defense Initiative at the Mucosal Surface. Annu. Rev. Immunol. 1986, 4, 389–417.
- Li, Y.; Jin, L.; Chen, T. The Effects of Secretory IgA in the Mucosal Immune System. BioMed Res. Int. 2020, 2020, 2032057.
- Pilette, C.; Ouadrhiri, Y.; Godding, V.; Vaerman, J.-P.; Sibille, Y. Lung mucosal immunity: Immunoglobulin-A revisited. Eur. Respir. J. 2001, 18, 571–588.
- Conley, M.E.; Delacroix, D.L. Intravascular and Mucosal Immunoglobulin A: Two Separate but Related Systems of Immune Defense? Ann. Intern. Med. 1987, 106, 892.
- Burnett, D. Immunoglobulins in the lung. Thorax 1986, 41, 337–344.
- Reboldi, A.; Arnon, T.I.; Rodda, L.B.; Atakilit, A.; Sheppard, D.; Cyster, J.G. IgA production requires B cell interaction with subepithelial dendritic cells in Peyer’s patches. Science 2016, 352, aaf4822.
- Stumbles, P.A.; Upham, J.W.; Holt, P.G. Airway dendritic cells: Co-ordinators of immunological homeostasis and immunity in the respiratory tract. APMIS 2003, 111, 741–755.
- Turula, H.; Wobus, C.E. The Role of the Polymeric Immunoglobulin Receptor and Secretory Immunoglobulins during Mucosal Infection and Immunity. Viruses 2018, 10, 237.
- Asano, M.; Komiyama, K. Polymeric immunoglobulin receptor. J. Oral Sci. 2011, 53, 147–156.
- Mantis, N.J.; Rol, N.; Corthésy, B. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol. 2011, 4, 603–611.
- Johansen, F.-E.; Kaetzel, C. Regulation of the polymeric immunoglobulin receptor and IgA transport: New advances in environmental factors that stimulate pIgR expression and its role in mucosal immunity. Mucosal Immunol. 2011, 4, 598–602.
- Gohy, S.T. Polymeric immunoglobulin receptor down-regulation in chronic obstructive pulmonary disease. Persistence in the cultured epithelium and role of transforming growth factor-beta. Am. J. Respir Crit. Care Med. 2014, 190, 509–521.
- Loman, S.; Jansen, H.M.; Out, T.A.; Lutter, R. Interleukin-4 and interferon-gamma synergistically increase secretory component gene expression, but are additive in stimulating secretory immunoglobulin A release by Calu-3 airway epithelial cells. Immunology 1999, 96, 537.
- Ladjemi, M.Z.; Gras, D.; Dupasquier, S.; Detry, B.; Lecocq, M.; Garulli, C.; Fregimilicka, C.; Bouzin, C.; Gohy, S.; Chanez, P. Bronchial Epithelial IgA Secretion Is Impaired in Asthma. Role of IL-4/IL-13. Am. J. Res-Piratory Crit. Care Med. 2018, 197, 1396–1409.
- Ratajczak, C.; Guisset, A.; Detry, B.; Sibille, Y.; Pilette, C. Dual effect of neutrophils on pIgR/secretory component in human bronchial epithelial cells: Role of TGF-beta. J. Biomed. Biotechnol. 2010, 2010, 428618.
- Collin, A.M.; Lecocq, M.; Noel, S.; Detry, B.; Carlier, F.M.; Nana, F.A.; Bouzin, C.; Leal, T.; Vermeersch, M.; De Rose, V.; et al. Lung immunoglobulin A immunity dysregulation in cystic fibrosis. eBioMedicine 2020, 60, 102947.
- Gohy, S.; Moeremans, A.; Pilette, C.; Collin, A. Immunoglobulin A Mucosal Immunity and Altered Respiratory Epithelium in Cystic Fibrosis. Cells 2021, 10, 3603.
- Carlier, F.M.; Detry, B.; Lecocq, M.; Collin, A.M.; Verleden, S.E.; Stanciu-Pop, C.M.; Janssens, W.; Ambroise, J.; Vanaudenaerde, B.M.; Gohy, S.T. The memory of airway epithelium damage in smokers and COPD patients. bioRxiv 2021.
- Guo, M.-Y.; Chen, H.-K.; Ying, H.-Z.; Qiu, F.-S.; Wu, J.-Q. The Role of Respiratory Flora in the Pathogenesis of Chronic Respiratory Diseases. BioMed Res. Int. 2021, 2021, 6431862.
- Sato, S.; Kiyono, H. The mucosal immune system of the respiratory tract. Curr. Opin. Virol. 2012, 2, 225–232.
- Carlier, F.M.; de Fays, C.; Pilette, C. Epithelial Barrier Dysfunction in Chronic Respiratory Diseases. Front. Physiol. 2021, 12, 691227.
- Vos, T.; Lim, S.S.; Abbafati, C.; Abbas, K.M.; Abbasi, M.; Abbasifard, M.; Abbasi-Kangevari, M.; Abbastabar, H.; Abd-Allah, F.; Abdelalim, A. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222.
- Salvi, S.S.; Barnes, P.J. Chronic obstructive pulmonary disease in non-smokers. Lancet 2009, 374, 733–743.
- Pilette, C.; Godding, V.; Kiss, R.; Delos, M.; Verbeken, E.; Decaestecker, C.; De Paepe, K.; Vaerman, J.-P.; Decramer, M.; Sibille, Y. Reduced Epithelial Expression of Secretory Component in Small Airways Correlates with Airflow Obstruction in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2001, 163, 185–194.
- Zuo, W.-L.; Rostami, M.R.; Shenoy, S.A.; LeBlanc, M.G.; Salit, J.; Strulovici-Barel, Y.; O’Beirne, S.L.; Kaner, R.J.; Leopold, P.L.; Mezey, J.G.; et al. Cell-specific expression of lung disease risk-related genes in the human small airway epithelium. Respir. Res. 2020, 21, 200.
- Polosukhin, V.V.; Cates, J.M.; Lawson, W.E.; Zaynagetdinov, R.; Milstone, A.P.; Massion, P.P.; Ocak, S.; Ware, L.B.; Lee, J.W.; Bowler, R.P. Bronchial secretory immunoglobulin a deficiency correlates with airway inflammation and progression of chronic obstructive pulmonary disease. Am. J. Respir Crit. Care Med. 2011, 184, 317–327.
- Polosukhin, V.V.; Richmond, B.W.; Du, R.-H.; Cates, J.; Wu, P.; Nian, H.; Massion, P.P.; Ware, L.B.; Lee, J.W.; Kononov, A.; et al. Secretory IgA Deficiency in Individual Small Airways Is Associated with Persistent Inflammation and Remodeling. Am. J. Respir. Crit. Care Med. 2017, 195, 1010–1021.
- Kuruvilla, M.E.; Lee, F.E.-H.; Lee, G.B. Understanding Asthma Phenotypes, Endotypes, and Mechanisms of Disease. Clin. Rev. Allergy Immunol. 2019, 56, 219–233.
- Wenzel, S.E. Asthma phenotypes: The evolution from clinical to molecular approaches. Nat. Med. 2012, 18, 716–725.
- Alhamwe, B.A.; Miethe, S.; Von Strandmann, E.P.; Potaczek, D.P.; Garn, H. Epigenetic Regulation of Airway Epithelium Immune Functions in Asthma. Front. Immunol. 2020, 11, 1747.
- Peters, U.; Dixon, A.E.; Forno, E. Obesity and asthma. J. Allergy Clin. Immunol. 2018, 141, 1169–1179.
- Potaczek, D.P.; Miethe, S.; Schindler, V.; Alhamdan, F.; Garn, H. Role of airway epithelial cells in the development of different asthma phenotypes. Cell. Signal. 2020, 69, 109523.
- Frey, A.; Lunding, L.P.; Ehlers, J.C.; Weckmann, M.; Zissler, U.M.; Wegmann, M. More than Just a Barrier: The Immune Functions of the Airway Epithelium in Asthma Pathogenesis. Front. Immunol. 2020, 11, 761.
- Nahm, D.-H.; Kim, H.-Y.; Park, H.-S. Elevation of specific immunoglobulin A antibodies to both allergen and bacterial antigen in induced sputum from asthmatics. Eur. Respir. J. 1998, 12, 540–545.
- Peebles, R.S.; Hamilton, R.G.; Lichtenstein, L.M.; Schlosberg, M.; Liu, M.C.; Proud, D.; Togias, A. Antigen-specific IgE and IgA antibodies in bronchoalveolar lavage fluid are associated with stronger antigen-induced late phase reactions. Clin. Exp. Allergy 2001, 31, 239–248.
- Orivuori, L.; Loss, G.; Roduit, C.; Dalphin, J.C.; Depner, M.; Genuneit, J.; Lauener, R.; Pekkanen, J.; Pfefferle, P.; Riedler, J. Soluble immunoglobulin A in breast milk is inversely associated with atopic dermatitis at early age: The PASTURE cohort study. Clin. Exp. Allergy 2014, 44, 102–112.
- Schaffer, F.M.; Monteiro, R.C.; Volanakis, J.E.; Cooper, M.D. IgA deficiency. Immunodefic. Rev. 1991, 3, 15–44.
- Kim, W.-J.; Choi, I.S.; Kim, C.S.; Lee, J.-H.; Kang, H.-W. Relationship between serum IgA level and allergy/asthma. Korean J. Intern. Med. 2017, 32, 137–145.
- Orivuori, L.; Mustonen, K.; Roduit, C.; Braun-Fahrländer, C.; Dalphin, J.-C.; Genuneit, J.; Lauener, R.; Pfefferle, P.; Riedler, J.; Weber, J.; et al. Immunoglobulin A and immunoglobulin G antibodies against β-lactoglobulin and gliadin at age 1 associate with immunoglobulin E sensitization at age 6. Pediatric Allergy Immunol. 2014, 25, 329–337.
- Hupin, C.; Rombaux, P.; Bowen, H.; Gould, H.; Lecocq, M.; Pilette, C. Downregulation of polymeric immunoglobulin receptor and secretory IgA antibodies in eosinophilic upper airway diseases. Allergy 2013, 68, 1589–1597.
- Ratjen, F.; Bell, S.C.; Rowe, S.M.; Goss, C.H.; Quittner, A.L.; Bush, A. Cystic fibrosis. Nat. Rev. Dis. Prim. 2015, 1, 15010.
- Collin, A.M.; Lecocq, M.; Detry, B.; Carlier, F.M.; Bouzin, C.; de Sany, P.; Hoton, D.; Verleden, S.; Froidure, A.; Pilette, C.; et al. Loss of ciliated cells and altered airway epithelial integrity in cystic fibrosis. J. Cyst. Fibros. 2021, 20, e129–e139.
- Marshall, L.J.; Perks, B.; Bodey, K.; Suri, R.; Bush, A.; Shute, J.K. Free Secretory Component from Cystic Fibrosis Sputa Displays the Cystic Fibrosis Glycosylation Phenotype. Am. J. Respir. Crit. Care Med. 2004, 169, 399–406.
- Hodson, M.E.; Morris, L.; Batten, J.C. Serum immunoglobulins and immunoglobulin G subclasses in cystic fibrosis related to the clinical state of the patient. Eur. Respir. J. 1988, 1, 701–705.
- Hassan, J.; Feighery, C.; Bresnihan, B.; Keogan, M.; Fitzgerald, M.X.; Whelan, A. Serum IgA and IGg Subclasses during Treatment for Acute Respiratory Exacerbation in Cystic Fibrosis: Analysis of Patients Colonised with Mucoid or Non-Mucoid Strains of Pseudomonas Aeruginosa. Immunol. Investig. 1994, 23, 1–13.
- Van Bever, H.P.; Gigase, P.L.; De Clerck, L.S.; Bridts, C.H.; Franckx, H.; Stevens, W.J. Immune complexes and Pseudomonas aeruginosa antibodies in cystic fibrosis. Arch. Dis. Child. 1988, 63, 1222–1228.
- Konstan, M.W.; Hilliard, K.A.; Norvell, T.M.; Berger, M. Bronchoalveolar lavage findings in cystic fibrosis patients with stable, clinically mild lung disease suggest ongoing infection and inflammation. Am. J. Respir Crit. Care Med. 1994, 150, 448–454.
- Verleden, G.M.; Raghu, G.; Meyer, K.C.; Glanville, A.R.; Corris, P. A new classification system for chronic lung allograft dysfunction. J. Hear. Lung Transplant 2014, 33, 127–133.
- Verleden, G.M.; Glanville, A.R.; Lease, E.D.; Fisher, A.J.; Calabrese, F.; Corris, P.A.; Ensor, C.R.; Gottlieb, J.; Hachem, R.R.; Lama, V. Chronic lung allograft dysfunction: Definition, diagnostic criteria, and approaches to treatment-A consensus report from the Pulmonary Council of the ISHLT. J. Heart Lung Transplant 2019, 38, 493–503.
- Martinu, T.; Howell, D.N.; Davis, R.D.; Steele, M.P.; Palmer, S.M. Pathologic correlates of bronchiolitis obliterans syn-drome in pulmonary retransplant recipients. Chest 2006, 129, 1016–1023.
- Le Pavec, J.; Pradère, P.; Gigandon, A.; Dauriat, G.; Dureault, A.; Aguilar, C.; Henry, B.; Lanternier, F.; Savale, L.; Dolidon, S.; et al. Risk of Lung Allograft Dysfunction Associated With Aspergillus Infection. Transplant. Direct 2021, 7, e675.
- Fisher, C.E.; Preiksaitis, C.M.; Lease, E.D.; Edelman, J.; Kirby, K.A.; Leisenring, W.M.; Raghu, G.; Boeckh, M.; Limaye, A.P. Symptomatic Respiratory Virus Infection and Chronic Lung Allograft Dysfunction. Clin. Infect. Dis. 2015, 62, 313–319.
- Gregson, A.L. Infectious Triggers of Chronic Lung Allograft Dysfunction. Curr. Infect. Dis. Rep. 2016, 18, 21.
- Bastian, A.; Tunkel, C.; Lins, M.; Bottcher, H.; Hirt, S.W.; Cremer, J.; Bewig, B. Immunoglobulin A and secretory immunoglobulin A in the bronchoalveolar lavage from patients after lung transplantation. Clin. Transplant. 2000, 14, 580–585.
- Vandermeulen, E.; Verleden, S.E.; Bellon, H.; Ruttens, D.; Lammertyn, E.; Claes, S.; Vandooren, J.; Ugarte-Berzal, E.; Schols, D.; Emonds, M.-P.; et al. Humoral immunity in phenotypes of chronic lung allograft dysfunction: A broncho-alveolar lavage fluid analysis. Transpl. Immunol. 2016, 38, 27–32.
- Murthy, S.C.; Avery, R.K.; Budev, M.; Gupta, S.; Pettersson, G.B.; Nowicki, E.R.; Mehta, A.; Chapman, J.T.; Rajeswaran, J.; Blackstone, E.H. Low pretransplant IgA level is associated with early post–lung transplant seromucous infection. J. Thorac. Cardiovasc. Surg. 2018, 156, 882–891.e8.
- Chambers, D.C.; Davies, B.; Mathews, A.; Yerkovich, S.T.; Hopkins, P.M. Bronchiolitis obliterans syndrome, hypogammaglobulinemia, and infectious complications of lung transplantation. J. Hear. Lung Transplant. 2013, 32, 36–43.
More