Ferroptosis, which has been widely associated with many diseases, is an iron-dependent regulated cell death characterized by intracellular lipid peroxide accumulation. The term ‘ferroptosis’, first proposed in 2012, refers to a programmed cell death resulting from iron-dependent lipid peroxidation accumulation. Ferroptosis is distinct from other previously established regulated cell deaths and has specific morphological, biochemical, and genetic characteristics. It exhibits morphological, biochemical, and genetic characteristics that are unique in comparison to other types of cell death. The course of ferroptosis can be accurately regulated by the metabolism of iron, lipids, amino acids, and various signal pathways. In this review, we summarize the basic characteristics of ferroptosis, its regulation, as well as the relationship between ferroptosis and chronic diseases such as cancer, nervous system diseases, metabolic diseases, and inflammatory bowel diseases. Finally, we describe the regulatory effects of food-borne active ingredients on ferroptosis.
- ferroptosis
- basic characteristics of ferroptosis
- regulation of ferroptosis
- chronic diseases
- foodborne active ingredients
1. Introduction
2. Basic Characteristics of Ferroptosis
2.1. Morphological Features
Ferroptosis has morphological, biochemical, and genetic features that are unique in comparison with apoptosis, autophagy, necroptosis, and pyroptosis (Table 1) [1,2,3][1][2][3]. Cells that undergo ferroptosis generally show a necrosis-like morphological transformation, including cell membrane rupture, cytoplasmic swelling, and moderate chromatin condensation [4]. At the ultrastructural level, ferroptotic cells usually present mitochondrial shrinkage, an increase in membrane density, reduced or absent cristae, and rupturing of the outer membrane [2]. Since autophagy promotes ferroptosis, autophagy-related ultrastructures, such as double-membrane autophagosomes and various lysosome-related vesicles, are often seen in ferroptotic cells or tissue [5].| Ferroptosis | Apoptosis | Autophagy | Necroptosis | Pyroptosis | |
|---|---|---|---|---|---|
| Morphological features | Mitochondrial shrinkage with increased mitochondrial membrane densities, reduced or vanished mitochondria crista, rupture of outer mitochondrial membrane | Plasma membrane blebbing, cellular and nuclear volume reduction, rounding-up of the cell, nuclear fragmentation, chromatin condensation | Formation of double-membraned autolysosomes | Rupture of plasma membrane, generalized swelling of the cytoplasm and organelles, moderate chromatin condensation | Karyopyknosis, cell edema and membrane rupture |
| Biochemical features | Iron accumulation and lipid peroxidation | DNA fragmentation | Increased lysosomal activity | Drop in ATP levels | Dependent on caspase-1 and proinflammatory cytokine releases |
| Genetic features | Positive: PTGS2, ACSL4, TFR1, NCOA4; Negative: GPX4, SLC7A11, NRF2, FSP1, FTH1 |
Positive: Bax, Bak, Bad, Bim, Bid; Negative: Bcl-2, Bcl-XL, Mcl-1 |
Positive: ATG5, ATG7, LC3, Beclin-1 | Positive: RIP1, RIP3, MLKL | Positive: Caspase-1, IL-1β, IL-18 |
2.2. Biochemical Features
The two main biochemical features of ferroptosis are iron accumulation and lipid peroxidation. Intracellular free ferrous iron accumulation generates excessive reactive oxygen species (ROS) directly via the Fenton reaction, thereby triggering lipid peroxidation and ferroptosis. Erastin induces ferroptosis by decreasing ferroportin (FPN) and increasing the level of ferrous ions (Fe2+) in endometriosis [6]. ROS-mediated autophagy results in ferroptosis via the degradation of ferritin and inducement of transferrin receptor (TFR1) expression to increase intracellular iron levels [7]. Iron overload in the motor cortex has been reported to trigger the ferroptosis of neuronal cells after spinal cord injury [8]. Ferroptosis inhibition attenuates atherosclerosis via the alleviation of lipid peroxidation [9].2.3. Genetic Features
Some genes are overexpressed in ferroptosis. For example, prostaglandin-endoperoxide synthase 2 (PTGS2/COX2) is known to play an important role in prostaglandin biosynthesis [10], while Acyl-CoA synthetase long-chain family member 4 (ACSL4) is an enzyme that increases the polyunsaturated fatty acid (PUFA) content in phospholipids, making them more sensitive to ferroptosis [11]. Other genes, such as glutathione peroxidase 4 (GPX4) and ferritin heavy chain 1 (FTH1), are downregulated in ferroptosis. GPX4 converts the cytotoxic lipid peroxides (L-OOH) into the corresponding alcohols (L-OH) under glutathione (GSH), and the suppression of GPX4 activity leads to lipid peroxides accumulation and ferroptosis. FTH1 is an iron-storage protein and, therefore, the reduction of its expression facilitates iron accumulation and ferroptosis.3. Regulation of Ferroptosis
Ferroptosis regulatory pathways are shown in Figure 1.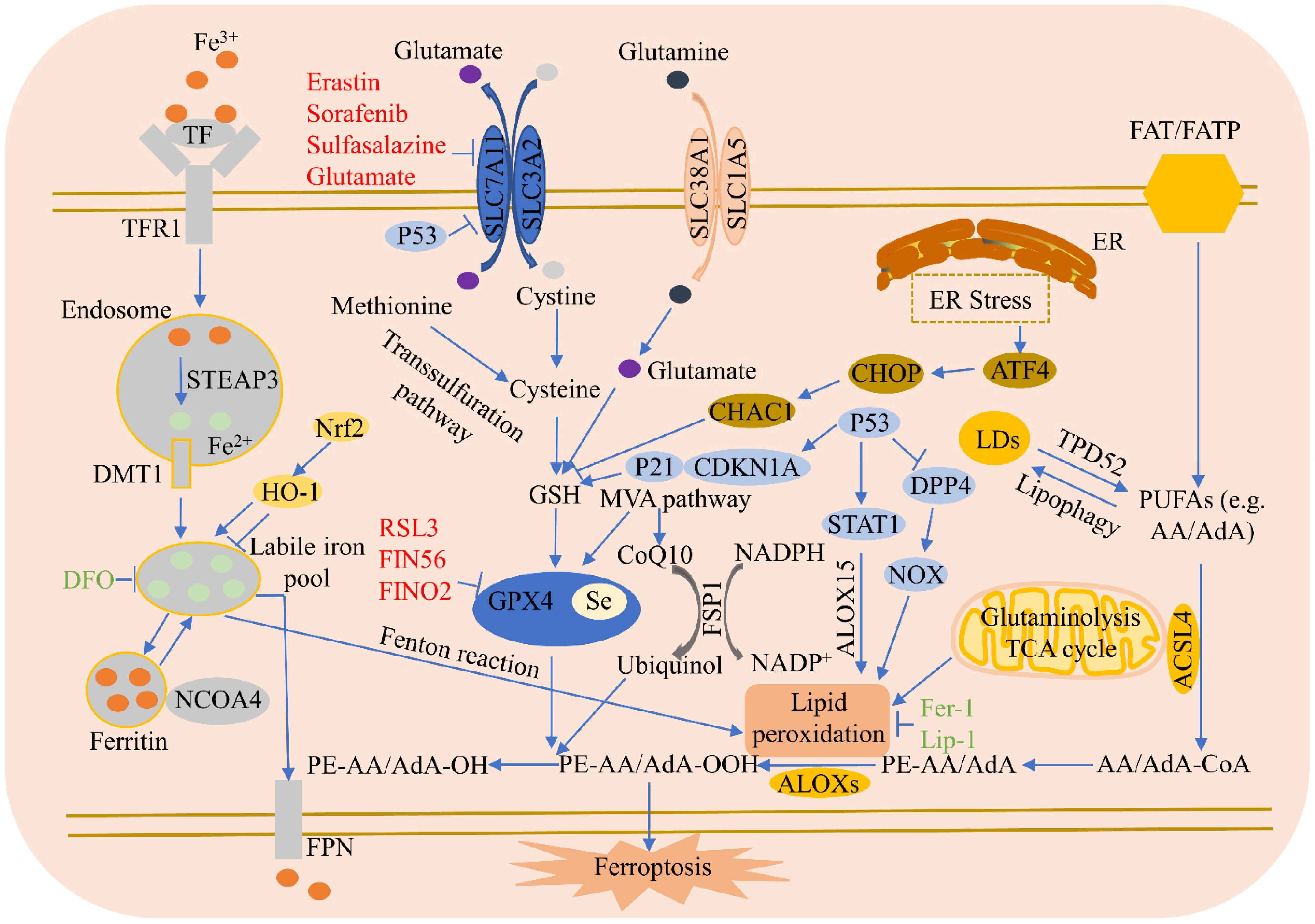
3.1. Iron Metabolism
Iron in food is principally Fe3+, which combines with transferrin (TF) in serum and is then identified by TFR1 in the cell membrane. Once absorbed by TFR1, Fe3+ is reduced to Fe2+ through the STEAP3 metalloreductase in the endosome, which is subsequently released from the endosome into the cytosol via solute carrier family 11 member 2 (SLC11A2/DMT1) [4]. The iron-storage protein ferritin plays an antiferroptosis role, including those of ferritin light chain and FTH1, and lysosomes can degrade ferritin to increase free iron levels. NCOA4-mediated ferritinophagy increases the degradation of ferritin by lysosomes, reduces iron storage, and promotes ferroptosis [12]. Finally, the iron-efflux protein FPN exports iron out of cells. Blocking the iron release pathway in cell membranes increases sensitivity to ferroptosis.3.2. Lipid Metabolism
The absorption of free PUFAs such as arachidonic acid (AA) or adrenoxyl (AdA) is mediated by fatty acid translocase (FAT) and fatty acid transport protein (FATP) [13]. ACSL4 facilitates the incorporation of PUFAs into cell membranes and ferroptosis. Lipoxygenases (ALOXs) mediate lipid peroxidation and cause ferroptosis. Tumor protein D52 (TPD52)-mediated lipid storage represses RSL3-induced lipid peroxidation and succedent ferroptosis [14]. Lipophagy, a process of autophagy, degrades lipid droplets (LDs), enhances the production of free fatty acids, and increases lipid peroxidation and ferroptosis [14]. Sterol carrier protein 2 promotes the transportation of peroxidized lipids to mitochondria and accelerates GPX4 depletion-mediated ferroptosis. Finally, acetyl-CoA carboxylase alpha is involved in fatty acid β-oxidation and biosynthesis, and promotes ferroptosis.3.3. Amino Acid Metabolism
Amino acid metabolism is closely related to ferroptosis regulation. Cysteine availability restricts GSH biosynthesis, while cysteine starvation induces GSH depletion and ferroptosis. When the available cysteine is limited, some cells utilize the transsulfuration pathway to transform methionine to cysteine [15]. These cells do not require the cystine/glutamate antiporter system Xc- to import cystine and, therefore, are resistant to system Xc- inhibitor-induced ferroptosis. Glutamate is also an important regulator of ferroptosis. In high concentrations, it suppresses system Xc- and triggers ferroptosis. Glutamine degradation (via glutaminolysis) fuels the tricarboxylic acid (TCA) cycle and provides the basis for necessary biosynthetic processes, such as lipid biosynthesis. Cystine starvation and blocked cystine uptake cannot increase ROS accumulation, lipid peroxidation, and ferroptosis in the presence of glutamine deficiency or the inhibition of glutaminolysis [16].3.4. System Xc-
The amino acid antiporter system Xc- consists of two subunits: SLC7A11 and SLC3A2. System Xc- imports extracellular cystine into cells by exchanging intracellular glutamate. Erastin, sorafenib, sulfasalazine, and glutamate all inhibit SLC7A11 expression, causing cysteine deprivation, GSH deletion, and GPX4 inactivation to trigger ferroptosis [17]. ATF3 enhances the ferroptosis induced by erastin via the repression of system Xc- [18], while AMPK-mediated BECN1 phosphorylation increases ferroptosis by directly inhibiting system Xc- activity [19]. Radiotherapy and immunotherapy enhance lipid oxidation and the ferroptosis of tumor cells by synergistically suppressing SLC7A11 [20]. Sorafenib inhibits system Xc- function and induces ferroptosis [21], while GDF15 knockdown facilitates the ferroptosis induced by erastin via the attenuation of SLC7A11 expression [22]. Cardiac ferritin H deficiency reduces SLC7A11 expression and facilitates ferroptosis and cardiomyopathy [23]. Nrf2 suppresses ferroptosis and reduces intestinal ischemia/reperfusion-induced acute lung injury (IIR-ALI) by increasing SLC7A11 and HO-1 [24]. PARP inhibition increases ferroptosis by inhibiting SLC7A11 and cooperates with ferroptosis inducers in BRCA-proficient ovarian cancer [25], while Nrf2 and STAT3 reduce ferroptosis by regulating SLC7A11, which alleviates IIR-ALI [26].3.5. GPX4
RSL3 represses GPX4 activity via covalent bonding with GPX4, resulting in the accumulation of lipid peroxides and, ultimately, ferroptosis. GSH is a co-factor in the catalysis of peroxides by GPX4 to produce alcohols; however, cysteine deprivation leads to GSH depletion, which directly inactivates GPX4, resulting in the subsequent induction of ferroptosis. Legumain has been shown to facilitate tubular ferroptosis via the promotion of chaperone-mediated GPX4 autophagy in acute kidney injury [27], while the selenium-GPX4 axis reduces the ferroptosis of follicular helper T cells [28]. Selenium is necessary for GPX4 to suppress hydroperoxide-induced ferroptosis [29,30][29][30]. FINO2 promotes ferroptosis through GPX4 inactivation and iron oxidation [31]. The downregulation of GPX4 during myocardial infarction triggers ferroptosis in cardiomyocytes [32]. Dihydroartemisinin (DHA) induces ferroptosis in glioblastoma through the inhibition of GPX4 [33]. SIRT3-activated autophagy promotes ferroptosis by increasing the AMPK/mTOR pathway and reducing GPX4 levels [34]. GPX4 maintains Treg cell activation and reduces antitumor immunity by inhibiting lipid peroxidation and ferroptosis [35]. Kaempferol protects from oxygen-glucose deprivation/reoxygenation-induced neuronal ferroptosis via the upregulation of the Nrf2/SLC7A11/GPX4 axis [36]. Finally, the inactivation of GPX4 was found to trigger ferroptosis and acute renal failure in mice [37].3.6. Ferroptosis Suppressor Protein 1 (FSP1)
FSP1 suppresses ferroptosis independent of GSH. Under nicotinamide adenine dinucleotide phosphate (NADPH), FSP1 reduces ubiquinone, also called coenzyme Q10 (CoQ10), to ubiquinol, which can reduce lipid peroxidation and succedent ferroptosis. The FSP1-CoQ10-NAD(P)H pathway has been shown to synergize with GPX4 and GSH to repress phospholipid peroxidation and ferroptosis [38], while a small molecule, NPD4928, reportedly enhances ferroptosis via the inhibition of FSP1 [39]. Plasma-activated medium promotes ferroptosis by decreasing FSP1 expression in human lung cancer cells [40]. Mesenchymal stem cell derived exosomes inhibit neuron ferroptosis by lncGm36569/microRNA (miR)-5627-5p/FSP1 axis in acute spinal cord injury [41], and MiR-672-3p inhibition facilitates neural recovery via the suppression of ferroptosis by FSP1 [42].3.7. P53
P53 is a tumor-suppressor gene that plays a dual role in ferroptosis, regulating it via either a transcriptional or post-translational mechanism. P53 decreases cystine absorption by transcriptionally suppressing SLC7A11 expression, reduces intracellular GSH, and induces ferroptosis in tumor cells [43]. P53 also enhances ferroptosis via the transcriptional induction of SAT1 or GSL2 [44]. On the other hand, p53 inhibits ferroptosis by transcriptionally inducing CDKN1A/p21 (cyclin-dependent kinase inhibitor 1 A) expression [45]. P53 also limits erastin-induced ferroptosis by blocking dipeptidyl peptidase 4 (DPP4) activity via a transcription-independent mechanism [46].3.8. Heme Oxygenase-1 (HO-1)
HO-1 plays a dual role in ferroptosis induction, associated with both the environment and cell type. HO-1 promotes the ferroptosis induced by erastin in HT-1080 fibrosarcoma cells [47], while tagitinin C promotes colorectal cancer cell ferroptosis by activating the PERK-Nrf2-HO-1 signaling pathway [48]. Ferroptosis inhibition effectively reduces dextran sulfate sodium (DSS)-induced ulcerative colitis, related to Nrf2/HO-1 signaling pathway blocking [49]. However, HO-1 also functions as a negative regulator of ferroptosis, and cetuximab enhances RSL3-induced ferroptosis in KRAS mutant colorectal cancer cells through the inhibition of the Nrf2/HO-1 pathway [50]. Gastrodin alleviates the ferroptosis of HT-22 cells triggered by glutamate by up-regulating the Nrf2/HO-1 signaling pathway [51]. HO-1 plays a key antiferroptotic role in renal epithelial cells [52]. Fraxetin decreases myocardial-infarction-mediated ferroptosis through AKT/Nrf2/HO-1 pathway activation [53]. MiR-3587 inhibitor promotes HO-1 up-regulation, thereby protecting renal tissues from ischemia/reperfusion-induced ferroptosis [54]. Melatonin reduces the level of ferroptosis by activating the Nrf2/HO-1signaling pathway in type 2 diabetic osteoporosis [55]. Panaxydol ameliorates lipopolysaccharide (LPS)-induced acute lung injury via reducing ferroptosis by upregulating the Keap1-Nrf2/HO-1 pathway [56]. Ginkgetin, which is derived from Ginkgo biloba leaves, increases cisplatin-induced ferroptosis via the disruption of the Nrf2/HO-1 axis [57].3.9. NCOA4
Recent studies have shown that ferritinophagy, a selective autophagy, can degrade ferritin. NCOA4, a selective cargo receptor, transports ferritin to lysosomes where it is then degraded, then labile iron is released and oxygen radicals are increased to induce ferroptosis [12]. NCOA4-mediated ferritinophagy enhances erastin-induced ferroptosis in HeLa cells [58]. The loss of coatomer protein complex subunit zeta 1 increases NCOA4 expression and the ferroptosis of glioblastoma cell lines [59]. DNA (cytosine-5)-methyltransferase 1 inhibition reduces ferroptosis via NCOA4-mediated ferritinophagy during diabetes myocardial ischemia/reperfusion injury [60]. Formosanin C promotes the ferroptosis of human hepatocellular carcinoma cells by elevating NCOA4-mediated ferritinophagy [61]. Disrupting NCOA4-FTH1 interaction inhibits ferroptosis [62]. NCOA4-mediated ferritinophagy increases the ferroptosis induced by cerebral ischemia [63].3.10. Endoplasmic Reticulum (ER) Stress
ER stress is triggered under different pathological conditions and is closely associated with the course of cell death. Erastin can induce ER stress and up-regulates ER-stress-responsive genes [21]. The eif2α-ATF4 axis is the ferroptotic reagent-activated primary signaling pathway. CHAC 1 is downstream of ATF4 and degrades GSH while promoting the succedent ferroptosis [64].3.11. Common Inducers and Inhibitors of Ferroptosis
The four known mechanisms of ferroptosis inducers are: (1) the inhibition of system Xc- and depletion of intracellular GSH by, for example, erastin, sorafenib, sulfasalazine, or glutamate; (2) the inhibition of GPX4 by, for example, RSL3, which covalently combines with GPX4 to reduce its activity, resulting in the accumulation of toxic lipid peroxides and, ultimately, ferroptosis; (3) the degradation of GPX4 and exhaustion of antioxidant CoQ10 by, for example, FIN56; and (4) the direct oxidation of ferrous iron and lipidome, or indirect inactivation of GPX4 by, for example, FINO2 [31]. There are two mechanisms of ferroptosis inhibitors: (1) inhibiting the accumulation of iron, such as that of deferoxamine (DFO), which chelates iron and limits the Fenton reaction to prevent lipid peroxidation; and (2) the inhibition of lipid peroxidation by, for example, ferrostatin-1 (Fer-1), liproxststatin-1 (Lip-1), and vitamin E, which act as radical scavengers to reduce lipid peroxides and effectively block ferroptosis. DFO is commercially available as Desferal, an iron chelator clinically approved for the treatment of acute iron intoxication and chronic iron overload [65]. For the action mechanism, Desferal is a hexadentate molecule that is able to bind free plasma iron and excess iron within cells at a 1-to-1 ratio and then is excreted via the urine or bile [66].References
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379.
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88.
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34.
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125.
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162.
- Li, Y.; Zeng, X.; Lu, D.; Yin, M.; Shan, M.; Gao, Y. Erastin induces ferroptosis via ferroportin-mediated iron accumulation in endometriosis. Hum. Reprod. 2021, 36, 951–964.
- Park, E.; Chung, S.W. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019, 10, 822.
- Feng, Z.; Min, L.; Chen, H.; Deng, W.; Tan, M.; Liu, H.; Hou, J. Iron overload in the motor cortex induces neuronal ferroptosis following spinal cord injury. Redox Biol. 2021, 43, 101984.
- Bai, T.; Li, M.; Liu, Y.; Qiao, Z.; Wang, Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic. Biol. Med. 2020, 160, 92–102.
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331.
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343.
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.R.; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428.
- Jiang, M.; Qiao, M.; Zhao, C.; Deng, J.; Li, X.; Zhou, C. Targeting ferroptosis for cancer therapy: Exploring novel strategies from its mechanisms and role in cancers. Transl. Lung Cancer Res. 2020, 9, 1569–1584.
- Bai, Y.; Meng, L.; Han, L.; Jia, Y.; Zhao, Y.; Gao, H.; Kang, R.; Wang, X.; Tang, D.; Dai, E. Lipid storage and lipophagy regulates ferroptosis. Biochem. Biophys. Res. Commun. 2019, 508, 997–1003.
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2021, 17, 2054–2081.
- Stockwell, B.R.; Friedmann, A.J.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285.
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072.
- Wang, L.; Liu, Y.; Du, T.; Yang, H.; Lei, L.; Guo, M.; Ding, H.F.; Zhang, J.; Wang, H.; Chen, X.; et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc. Cell Death Differ. 2020, 27, 662–675.
- Song, X.; Zhu, S.; Chen, P.; Hou, W.; Wen, Q.; Liu, J.; Xie, Y.; Liu, J.; Klionsky, D.J.; Kroemer, G.; et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc- Activity. Curr. Biol. 2018, 28, 2388–2399.e5.
- Lang, X.; Green, M.D.; Wang, W.; Yu, J.; Choi, J.E.; Jiang, L.; Liao, P.; Zhou, J.; Zhang, Q.; Dow, A.; et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019, 9, 1673–1685.
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523.
- Chen, L.; Qiao, L.; Bian, Y.; Sun, X. GDF15 knockdown promotes erastin-induced ferroptosis by decreasing SLC7A11 expression. Biochem. Biophys. Res. Commun. 2020, 526, 293–299.
- Fang, X.; Cai, Z.; Wang, H.; Han, D.; Cheng, Q.; Zhang, P.; Gao, F.; Yu, Y.; Song, Z.; Wu, Q.; et al. Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ. Res. 2020, 127, 486–501.
- Dong, H.; Qiang, Z.; Chai, D.; Peng, J.; Xia, Y.; Hu, R.; Jiang, H. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging (Albany NY) 2020, 12, 12943–12959.
- Hong, T.; Lei, G.; Chen, X.; Li, H.; Zhang, X.; Wu, N.; Zhao, Y.; Zhang, Y.; Wang, J. PARP inhibition promotes ferroptosis via repressing SLC7A11 and synergizes with ferroptosis inducers in BRCA-proficient ovarian cancer. Redox Biol. 2021, 42, 101928.
- Qiang, Z.; Dong, H.; Xia, Y.; Chai, D.; Hu, R.; Jiang, H. Nrf2 and STAT3 Alleviates Ferroptosis-Mediated IIR-ALI by Regulating SLC7A11. Oxid. Med. Cell. Longev. 2020, 2020, 5146982.
- Chen, C.; Wang, D.; Yu, Y.; Zhao, T.; Min, N.; Wu, Y.; Kang, L.; Zhao, Y.; Du, L.; Zhang, M.; et al. Legumain promotes tubular ferroptosis by facilitating chaperone-mediated autophagy of GPX4 in AKI. Cell Death Dis. 2021, 12, 65.
- Yao, Y.; Chen, Z.; Zhang, H.; Chen, C.; Zeng, M.; Yunis, J.; Wei, Y.; Wan, Y.; Wang, N.; Zhou, M.; et al. Selenium-GPX4 axis protects follicular helper T cells from ferroptosis. Nat. Immunol. 2021, 22, 1127–1139.
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto, F.F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21.
- Conrad, M.; Proneth, B. Selenium: Tracing Another Essential Element of Ferroptotic Cell Death. Cell Chem Biol 2020, 27, 409–419.
- Gaschler, M.M.; Andia, A.A.; Liu, H.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018, 14, 507–515.
- Park, T.J.; Park, J.H.; Lee, G.S.; Lee, J.Y.; Shin, J.H.; Kim, M.W.; Kim, Y.S.; Kim, J.Y.; Oh, K.J.; Han, B.S.; et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 2019, 10, 835.
- Yi, R.; Wang, H.; Deng, C.; Wang, X.; Yao, L.; Niu, W.; Fei, M.; Zhaba, W. Dihydroartemisinin initiates ferroptosis in glioblastoma through GPX4 inhibition. Biosci. Rep. 2020, 40, BSR20193314.
- Han, D.; Jiang, L.; Gu, X.; Huang, S.; Pang, J.; Wu, Y.; Yin, J.; Wang, J. SIRT3 deficiency is resistant to autophagy-dependent ferroptosis by inhibiting the AMPK/mTOR pathway and promoting GPX4 levels. J. Cell. Physiol. 2020, 235, 8839–8851.
- Xu, C.; Sun, S.; Johnson, T.; Qi, R.; Zhang, S.; Zhang, J.; Yang, K. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep. 2021, 35, 109235.
- Yuan, Y.; Zhai, Y.; Chen, J.; Xu, X.; Wang, H. Kaempferol Ameliorates Oxygen-Glucose Deprivation/Reoxygenation-Induced Neuronal Ferroptosis by Activating Nrf2/SLC7A11/GPX4 Axis. Biomolecules 2021, 11, 923.
- Friedmann, A.J.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191.
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; Da, S.M.; Ingold, I.; Goya, G.A.; Xavier, D.S.T.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698.
- Yoshioka, H.; Kawamura, T.; Muroi, M.; Kondoh, Y.; Honda, K.; Kawatani, M.; Aono, H.; Waldmann, H.; Watanabe, N.; Osada, H. Identification of a Small Molecule That Enhances Ferroptosis via Inhibition of Ferroptosis Suppressor Protein 1 (FSP1). ACS Chem. Biol. 2022, 17, 483–491.
- Jo, A.; Bae, J.H.; Yoon, Y.J.; Chung, T.H.; Lee, E.W.; Kim, Y.H.; Joh, H.M.; Chung, J.W. Plasma-activated medium induces ferroptosis by depleting FSP1 in human lung cancer cells. Cell Death Dis. 2022, 13, 212.
- Shao, C.; Chen, Y.; Yang, T.; Zhao, H.; Li, D. Mesenchymal Stem Cell Derived Exosomes Suppress Neuronal Cell Ferroptosis Via lncGm36569/miR-5627-5p/FSP1 Axis in Acute Spinal Cord Injury. Stem. Cell Rev. Rep. 2022, 18, 1127–1142.
- Wang, F.; Li, J.; Zhao, Y.; Guo, D.; Liu, D.; Chang, S.; Qiao, H.; Li, J.; Yang, Y.; Zhang, C.; et al. miR-672-3p Promotes Functional Recovery in Rats with Contusive Spinal Cord Injury by Inhibiting Ferroptosis Suppressor Protein 1. Oxid. Med. Cell. Longev. 2022, 2022, 6041612.
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62.
- Kang, R.; Kroemer, G.; Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol. Med. 2019, 133, 162–168.
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575.
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704.
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403.
- Wei, R.; Zhao, Y.; Wang, J.; Yang, X.; Li, S.; Wang, Y.; Yang, X.; Fei, J.; Hao, X.; Zhao, Y.; et al. Tagitinin C induces ferroptosis through PERK-Nrf2-HO-1 signaling pathway in colorectal cancer cells. Int. J. Biol. Sci. 2021, 17, 2703–2717.
- Chen, Y.; Zhang, P.; Chen, W.; Chen, G. Ferroptosis mediated DSS-induced ulcerative colitis associated with Nrf2/HO-1 signaling pathway. Immunol. Lett. 2020, 225, 9–15.
- Yang, J.; Mo, J.; Dai, J.; Ye, C.; Cen, W.; Zheng, X.; Jiang, L.; Ye, L. Cetuximab promotes RSL3-induced ferroptosis by suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant colorectal cancer. Cell Death Dis. 2021, 12, 1079.
- Jiang, T.; Cheng, H.; Su, J.; Wang, X.; Wang, Q.; Chu, J.; Li, Q. Gastrodin protects against glutamate-induced ferroptosis in HT-22 cells through Nrf2/HO-1 signaling pathway. Toxicol. Vitr. 2020, 62, 104715.
- Adedoyin, O.; Boddu, R.; Traylor, A.; Lever, J.M.; Bolisetty, S.; George, J.F.; Agarwal, A. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am. J. Physiol. Renal Physiol. 2018, 314, F702–F714.
- Xu, Y.; Lin, H.; Wang, H.; Pang, J.; Zhou, Y. Fraxetin attenuates ferroptosis in myocardial infarction via AKT/Nrf2/HO-1 signaling. Am. J. Transl. Res. 2021, 13, 10315–10327.
- Tao, W.; Liu, F.; Zhang, J.; Fu, S.; Zhan, H.; Qian, K. miR-3587 Inhibitor Attenuates Ferroptosis Following Renal Ischemia-Reperfusion Through HO-1. Front. Mol. Biosci. 2021, 8, 789927.
- Ma, H.; Wang, X.; Zhang, W.; Li, H.; Zhao, W.; Sun, J.; Yang, M. Melatonin Suppresses Ferroptosis Induced by High Glucose via Activation of the Nrf2/HO-1 Signaling Pathway in Type 2 Diabetic Osteoporosis. Oxid. Med. Cell. Longev. 2020, 2020, 9067610.
- Li, J.; Lu, K.; Sun, F.; Tan, S.; Zhang, X.; Sheng, W.; Hao, W.; Liu, M.; Lv, W.; Han, W. Panaxydol attenuates ferroptosis against LPS-induced acute lung injury in mice by Keap1-Nrf2/HO-1 pathway. J. Transl. Med. 2021, 19, 96.
- Lou, J.S.; Zhao, L.P.; Huang, Z.H.; Chen, X.Y.; Xu, J.T.; Tai, W.C.; Tsim, K.; Chen, Y.T.; Xie, T. Ginkgetin derived from Ginkgo biloba leaves enhances the therapeutic effect of cisplatin via ferroptosis-mediated disruption of the Nrf2/HO-1 axis in EGFR wild-type non-small-cell lung cancer. Phytomedicine 2021, 80, 153370.
- Gryzik, M.; Asperti, M.; Denardo, A.; Arosio, P.; Poli, M. NCOA4-mediated ferritinophagy promotes ferroptosis induced by erastin, but not by RSL3 in HeLa cells. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118913.
- Zhang, Y.; Kong, Y.; Ma, Y.; Ni, S.; Wikerholmen, T.; Xi, K.; Zhao, F.; Zhao, Z.; Wang, J.; Huang, B.; et al. Loss of COPZ1 induces NCOA4 mediated autophagy and ferroptosis in glioblastoma cell lines. Oncogene 2021, 40, 1425–1439.
- Li, W.; Li, W.; Wang, Y.; Leng, Y.; Xia, Z. Inhibition of DNMT-1 alleviates ferroptosis through NCOA4 mediated ferritinophagy during diabetes myocardial ischemia/reperfusion injury. Cell Death Discov. 2021, 7, 267.
- Lin, P.L.; Tang, H.H.; Wu, S.Y.; Shaw, N.S.; Su, C.L. Saponin Formosanin C-induced Ferritinophagy and Ferroptosis in Human Hepatocellular Carcinoma Cells. Antioxidants 2020, 9, 682.
- Fang, Y.; Chen, X.; Tan, Q.; Zhou, H.; Xu, J.; Gu, Q. Inhibiting Ferroptosis through Disrupting the NCOA4-FTH1 Interaction: A New Mechanism of Action. ACS Cent. Sci. 2021, 7, 980–989.
- Li, C.; Sun, G.; Chen, B.; Xu, L.; Ye, Y.; He, J.; Bao, Z.; Zhao, P.; Miao, Z.; Zhao, L.; et al. Nuclear receptor coactivator 4-mediated ferritinophagy contributes to cerebral ischemia-induced ferroptosis in ischemic stroke. Pharmacol. Res. 2021, 174, 105933.
- Chen, M.S.; Wang, S.F.; Hsu, C.Y.; Yin, P.H.; Yeh, T.S.; Lee, H.C.; Tseng, L.M. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2α-ATF4 pathway. Oncotarget 2017, 8, 114588–114602.
- Yi, L.; Ju, Y.; He, Y.; Yin, X.; Xu, Y.; Weng, T. Intraperitoneal injection of Desferal® alleviated the age-related bone loss and senescence of bone marrow stromal cells in rats. Stem. Cell Res. Ther. 2021, 12, 45.
- Velasquez, J.; Wray, A.A. Deferoxamine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 8 May 2022.